Genetic Associations
How an international collaboration launched a new era of MS research
On the second Tuesday of every month, about 20 researchers from a dozen or so cities around the world get on the horn to participate in a conference call that has been happening regularly for nearly a decade. The sun is just peeking over the Australian skyline when participants dial in from Melbourne and Sydney. Their colleagues in Boston and other cities across the United States reach for their office phones during more reasonable work hours, while those in Norway, Finland, and Sweden might already be wearing pajamas. For the members of the International Multiple Sclerosis Genetics Consortium (IMSGC), the years of calls have held the group together, not just by keeping the far-flung collaborators informed of projects under way, but also by providing the means for would-be competitors to develop enough trust to share their hunches, their plans, and their data.
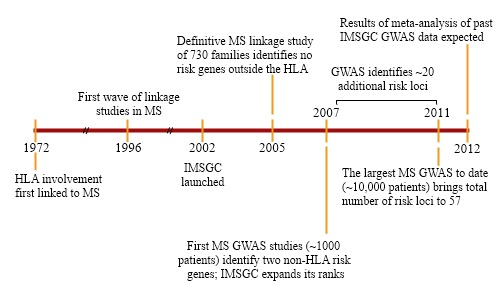
When the IMSGC formed, in 2002, the idea of competing labs pooling their resources—and of principal investigators forgoing primary authorship on research papers—was still novel. But researchers studying the genetics of MS were stumped. The malady clearly clustered in families; 15% to 20% of patients have a relative with the disease, and having a twin with MS confers a 30% risk of developing the illness. But the search for culprit genes had hit a dead end.
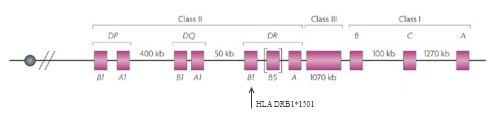
Only one risk locus, a particular region on chromosome 6 that is part of the major histocompatibility complex (MHC), had been solidly tied to MS—and the first reports of that connection had appeared 30 years earlier (Jersild et al., 1972). The MHC family of genes, called the human leukocyte antigen (HLA) system in humans, encodes proteins on the cell surface that present antigens—typically, small chunks of foreign or a person’s own proteins—to T cells. Among other things, this process helps the immune system distinguish invaders from its own components. Researchers first correlated certain HLAs with MS in the early 1970s and, as genotyping technology evolved, traced the bulk of the disease-risk effect to specific alleles, or variants, of the genes, most significantly HLA DRB1*1501 (Barcellos et al., 1996). This locus accounted for a sizable chunk of the genetic, as opposed to the environmental, component of risk for the disease. But most of the heritability remained elusive.
Although many genes outside the MHC had been proposed, each one failed to hold up when researchers attempted to replicate the findings, and the experts were beginning to realize why: The studies had too few patients to yield statistically significant results. Unlike disorders such as cystic fibrosis or Huntington disease, which stem from glitches in a single gene, MS arises from a combination of genetic and environmental factors. Each of the multiple genes involved exerts a small influence on whether someone develops the disease, as do a variety of environmental factors. Tracking this legion of modest effects required many more study subjects than scientists initially realized. “No one [research] group had the ability to gather together the requisite number of samples to make progress,” says Alastair Compston, a clinical neuroscientist at Cambridge University in the U.K., who had helped pin down the importance of the original MHC locus in MS.
That realization helped spur a collaborative effort that has cost about $25 million over the past decade and has charted today’s more detailed genetic landscape. A study published in August 2011, by far the biggest of its kind for MS, brought to 57 the number of spots on the human genome where variations were associated with increased risk of the disease. Since then, three more have been identified, and further follow-up work is poised to push the total number over 100 within the next year. Insights from such investigations have not yet helped people affected by this condition. (The same can be said for many other complex diseases.) “If the genetics [of MS] had been different, there might have been an effect on clinical practice,” says Jonathan Haines, a geneticist at Vanderbilt University in Nashville, Tennessee. But scientists involved in the endeavor say that pinpointing variants in the genome can outline molecular pathways that define the malady’s onset, and these pathways can be targeted when devising new therapies.
Even though it has taken decades to detect the variants, the hard part begins now, researchers say: identifying the genes that are associated with them, elucidating the mechanisms by which these genes make their carriers more susceptible to MS, and ultimately, translating this information to the clinic. It won’t be easy. Geneticists are still developing basic techniques for analyzing such studies; figuring out which gene corresponds to a variant is far from straightforward, and identifying the function of any novel gene can consume an entire career. “There’s a lot of work in front of us,” says Hanne Harbo, an MS geneticist at Oslo University Hospital in Norway.
Broken linkage, strong associations
The story of collaboration in the study of MS genetics stretches back well before the start of the IMSGC. By the 1990s, researchers were beginning to realize the limitations of going solo, and small-scale cooperative efforts were beginning to emerge, for example among researchers in Nordic countries or across Australia and New Zealand. Most gene-mining projects had deployed a technique called linkage analysis, in which researchers use well-defined sequences of DNA as markers to track how risk for MS is inherited within individual families. Markers close to the genes or sequences that stir up the biological trouble would be passed down with the miscreant DNA, pointing researchers to short stretches of the genome in which the culprits lie.
Linkage studies work well to identify mutations that play a large role in a disease. But in cases of complex inheritance, when many alleles contribute to disease risk, related family members are likely to have different assortments, making the relevance of each one harder to demonstrate statistically and rendering linkage studies ineffective. Based on a string of ambiguous and disappointing papers using the technique, MS was beginning to look like such a case (Ebers et al., 1996; Broadley et al., 2001; GAMES and Transatlantic Multiple Sclerosis Genetics Cooperative, 2003). A different approach was needed: association studies, which fish out disease-associated variants by comparing markers among large numbers of affected and unaffected individuals instead of comparing inheritance patterns within families. So-called genome-wide association studies (GWAS) scan hundreds of thousands of points along the genome and identify alleles that are more common in those who have the disease compared to those who do not.
In the late 1990s, Compston, at Cambridge, decided to get groups across Europe to pool their samples and expertise and to take the first serious stab at conducting an association study for MS. The collaborators called the effort GAMES (Genetic Analysis of Multiple Sclerosis in Europeans Consortium). A theoretical paper had suggested that a map of 6000 markers in 200 MS patients and 200 controls would provide sufficient statistical power (Barcellos et al., 1997). “We now know that you need to do hundreds of thousands of tests in thousands of individuals,” says Stephen Sawcer, a clinical neurologist also at Cambridge. The massive underestimate meant the study found no risk alleles. Still, GAMES paid off. “We did not succeed in discovering anything, but what we did achieve was the concept of working together,” Compston says. “The cultural dividend from GAMES was very high, even if the scientific dividend was low.”
At the time, David Hafler, an immunologist then studying MS at Harvard University, was thinking about the future of his field. “In the 1980s, there were two cytokines, IL1 and IL2, and CD4 and CD8 T cells,” he says. Because so few of the players were known, one could study them all. As knowledge about immunology exploded, showering the terrain with newly discovered molecules, he realized that he and his colleagues needed a scaffold for MS on which to hang these immune components. Furthermore, he thought that genomics could provide such a framework. He reached out to genomicist Eric Lander, then also at Harvard, to learn about complex genetics and genome sequencing. “When I met with Eric, I said, ‘What do I need to read?’ He said, ‘Well, nothing has been written yet,’ ” Hafler recalls.
A few groups of researchers, such as those who study diabetes and inflammatory bowel disease, had begun collaborating to tease apart the relevant genetics. By 2002, those founders of the IMSGC who had already been involved with genetics consortia dedicated to MS began to discuss how they could again work together; Hafler also began to chat with Stephen Hauser of the University of California, San Francisco, about the idea of a new international consortium for MS. At around this time, while at a conference in Copenhagen, Hafler and Compston continued those conversations with several colleagues including Haines and Margaret Pericak-Vance, now at the University of Miami in Florida. They envisioned an ambitious goal: discovering all of the common genetic risk variants in MS. “These were all the big competing groups,” says Hafler, who is now at Yale. All agreed that the strategy was promising, but it needed funding.
Hafler asked Adrian Ivinson, director of Harvard’s NeuroDiscovery Center, to help compose a proposal to raise money for the nascent group; the center has since acted as the administrative coordinator for the consortium as well as one of its many scientific partners. Initially, the team reached out to private philanthropists. A Boston venture capitalist named Martha Crowninshield, who has a long-standing interest in MS and has remained closely involved with the IMSGC’s work, helped them refine the proposal and contributed the first $1 million. She also hosted an event at which a handful of potential donors were invited to hear about the consortium’s plans from Hafler, Lander, and Joseph Martin, then the dean of Harvard Medical School. At evening’s end, the NeuroDiscovery Center had a $3 million commitment for the project—and the means to launch its scientific agenda.
Establishing group guidelines
Although the consortium members were eager to collaborate, Hafler says, “obviously, people have their egos.” When disagreements arose and discussions stalled, he recalls, “Steve [Hauser] and I would say to each other, ‘Oh my god, we’re getting old here. We started working on this problem in the early 1980s, and now it’s 30 years later.’” The initial crew members knew they would have to replace guarded competition with trust, so one of their first steps was to create a charter—appropriated and amended from the International Inflammatory Bowel Disease Genetics Consortium—that addressed issues such as authorship, seniority, sample and data sharing, bringing in new members, and settling disputes. Decisions would be made by group consensus, and members agreed to a “no surprises” rule. This policy was meant to ensure that the boundaries between IMSGC projects and individual labs’ work were known to everyone in advance. The founding members also agreed that the collaboration would stay small until time had proven that it could work. Additionally, one of the group’s guiding principles was consideration for young investigators; those participants in particular needed to receive credit for their work and to take the lead on ambitious projects with IMSGC data in order to build their own careers.
Not everyone was convinced that linkage studies for MS were a dead end. First on the group’s agenda, then, was a linkage study to end all linkage studies—that is, a linkage study powerful enough to definitively determine whether the technique could identify genetic risk factors outside the MHC. The answer to the question, based on a study of 700 families, turned out to be a resounding “no” (Sawcer et al., 2005). The analysis once again pointed to the HLA DRB1*1501 allele in the MHC, and it identified no other signals anywhere near as strong, despite being many orders of magnitude more sensitive than previous MS linkage studies.
Meanwhile, the technology that powers genetic research was on the verge of a revolution. In the 2000s, researchers were turning to single-nucleotide polymorphisms (SNPs)—single-base variations in the genome—which could serve as much denser, more accurate markers than those previously used. Initiatives sparked by the Human Genome Project, such as the SNP Consortium and the International HapMap Project, were identifying tens of thousands of SNPs common enough to be found in 1% to 5% of the population. This development meant that researchers could use a microchip that contained upward of 100,000 such SNPs to scan a person’s entire genome and assess which variants were more common in people with a specific disease.
With linkage a failure, the group was divided about how to proceed. Some members were eager to try GWAS in MS; however, the technique was costly and unproven, and others thought it more prudent to wait for the technology to mature. “We had no idea whether GWAS would work,” Hafler says, and as he recalls it, the 2004 Christmas holidays simmered with “many very painful conversations working through the issues.” Several weeks of back and forth convinced the consortium’s more conservatively minded individuals, Hafler says, and early the following year, the group embarked on a GWAS of 931 patients, with their unaffected parents serving as controls. A $2.6 million grant from the National Multiple Sclerosis Society that came through in mid-2005 provided the needed bolus to complete the project, and the resulting study, published in 2007 (IMSGC et al., 2007), broke the dry spell and fingered two genes outside the MHC—those that encode the alpha-chain components of the interleukin 7 receptor (IL7RA) and the interleukin 2 receptor (IL2RA)—as risk loci for the disease.
The findings marked a turning point because they showed that GWAS held power where linkage studies did not. Both genes, however, had been the focus of earlier MS research. IL7R had been a veritable whack-a-mole of MS genetics. Researchers linked its overactivity to the disease in a 2001 report of 15 patients with relapsing-remitting MS (Ramanathan et al., 2001), but subsequent studies of the gene were inconclusive. When IMSGC participants and others further investigated the gene, they found that the risk variant changes how it is spliced, which likely causes it to generate reduced amounts of the protein’s alpha chain in the membrane and increased amounts in the cytoplasm (Gregory et al., 2007). Because this portion of the protein must lie in the membrane for the receptor to work properly, the location change curbs its ability to do its job. Away from the membrane, it is no longer available to respond to a cytokine called IL-7, which normally helps certain lymphocytes survive and promotes development of B and T cells. Perhaps dampening T- and B-cell maturation could knock immune cell function off kilter and contribute to MS, researchers conjectured. The second gene identified in the association study, IL2RA, had also been implicated in MS (as well as other autoimmune disorders); an antibody therapy called daclizumab, which targets the protein, is now in phase III trials for the disease.
A mathematical analysis of the study, however, showed that it, too, lacked statistical power, suggesting that additional risk variants might be found with larger data sets. Researchers took a long time to understand how many subjects were needed to render GWAS useful, Sawcer says, in large part because the number required depends on the relative number of SNPs and risk genes—and on how strongly the genes involved boost the likelihood of getting the disease. “With 10,000,000 common variants, the chance that any one is relevant is very low,” he says, and it turns out that most genes that underlie a complex disorder such as MS raise the risk of the disease by less than a factor of 2. Taking these issues into consideration, investigators have now calculated that a minimum of 2000 cases and as many controls are needed to achieve statistical significance, Sawcer says—about twice as many individuals as were included in the 2007 study. Additional subjects would increase the study’s power even more.
As other susceptibility loci—most of them with unknown roles in the body—began to trickle in through subsequent studies, the group began planning a GWAS with 10 times as many patients. “We quickly determined that if we wanted to be able to collect that number of samples, we would have to expand the consortium,” Ivinson says.
Branching out
The IMSGC began by drawing groups from across Europe and Australia—those that had been collaborating informally via the GAMES connection—into the fold. It then invited others to join. “Because we already had a structure that worked, bringing in other people was easy,” Haines says. Several consortium members estimate that the IMSGC now comprises about 9 out of every 10 researchers working on MS genetics worldwide. (Unless otherwise specified, every scientist quoted in this story is a member of the group.)
Prospective members must be proposed by current consortium members. Candidates must bring something new to the group—almost always patient samples, although in some cases, expertise. “We’re not trying to capture [all researchers], and we don’t want this huge bureaucracy that slows things down,” Ivinson says, adding that the goal is to strike a balance between being open and being productive.
The fact that the IMSGC encompasses such a large swath of the MS genetics community is a tremendous boost for efforts aimed at identifying genes with small effects, says Brian Weinshenker, a neurologist at the Mayo Clinic in Rochester, Minnesota, who uses a different approach to study MS genetics and is not a member of the group—but it might not be entirely healthy for the field, he adds. With the majority of scientists involved, he says, few are left to provide what good science needs: constructive criticism from informed colleagues.
Some potential members have been turned away, Ivinson says, while others have balked at the notion of relinquishing their samples to be stored in the consortium’s two repositories, one maintained by the Cambridge group and the other by Pericak-Vance’s group at the University of Miami. That point is non-negotiable, he says. Going through written approvals and waiting for materials to be aliquotted, checked, and shipped from a home laboratory could easily add 6 months to an experiment. In contrast, the consortium repositories are geared to prep large numbers of samples for action as quickly as possible.
That setup helped the consortium to get moving on its latest GWAS, a $5 million to $6 million effort that included 9772 patients and 17,376 controls. It was conducted as part of the Wellcome Trust Case Control Consortium, a massive study of genetic variation in common diseases. The resulting report, published in August 2011, scanned about 600,000 SNPs—covering approximately 80% of the genome—and brought the total number of confirmed risk variants for MS to 57 (IMSGC et al., 2011). Furthermore, it pointed to an additional 50 or so SNPs that showed a signal but didn’t reach statistical significance; consortium members expect many of those hits to be confirmed in follow-up work, most notably a meta-analysis consisting of data from the latest GWAS and 11,000 additional cases that is now in progress.
From associations to insights
The GWAS findings won’t change clinical care anytime soon. “The purpose of these studies was not necessarily to provide information for patients but more of an effort to understand the basic biology that leads people to MS,” says Philip De Jager, an MS geneticist at Harvard Medical School and one of the leaders of the most recent GWAS work.
The slew of genes now linked to the disease provides the first coherent outline of its underlying genetic architecture, De Jager says. Because these features are present from before birth, he says, “the genetics are telling us about the earliest events in MS—events we were never really able to capture before.”
An initial analysis, in which researchers looked for associations between the risk genes and other aspects of the disease, such as its type and severity, suggested that these genes might play a role in whether a person gets the disease, but not in the disease’s course. If that’s the case, the genes might not prove to be good targets for new therapies, says Jan Hillert, a neurologist at the Karolinska Institute in Stockholm, Sweden, who started the Nordic MS genetics consortium in 1994 and joined the IMSGC about 4 years ago. He and others note, however, that they’ll need better ways of stratifying patients and more robust statistical methods to know for sure whether the genetic risk factors relate to anything about the disease other than risk.
The findings are also relevant to a key question in MS pathogenesis: whether MS starts when a neurodegenerative process incites inflammation or when an inflammatory process mistakenly targets the brain. The vast majority of genes identified in the study have immune functions, De Jager says, strongly supporting the latter explanation. In particular, many of the genes hit pathways that activate lymphocytes. But several researchers note that a neurodegenerative cause can’t yet be ruled out. Sergio Baranzini, an MS geneticist at the University of California, San Francisco, points out that tremendous overlap exists between genes that operate in different physiological processes, and that the so-called immune genes could also function in the nervous system. His group is discovering that the genes found by the GWAS are active there as well (Baranzini et al., 2009).
Information from the study is already dovetailing with other known aspects of the disease. Many of the genes with the strongest association to MS, including HLA DRB1*1501, appear to be regulated by vitamin D, says MS geneticist George Ebers of Oxford University in the U.K. (who is not a member of the IMSGC). Because vitamin D levels strongly depend on sunlight, the observation fits with epidemiological studies showing that lack of sun exposure is an important risk factor for the disease. “If this gene variance can explain how,” Harbo says, “that’s really a big step forward” (see "The Sunshine Suspect").
Many of the genes associated with MS have also been linked with related disorders, but not always in straightforward ways. A study published in December 2011, which identified three more variants in addition to the 57 found in the latest GWAS, also compared risk genes for MS with those for ulcerative colitis, diabetes, psoriasis, and other autoimmune diseases. It found that a sizable minority of risk genes shared with celiac disease and other conditions have an opposite effect—protective rather than injurious—to that in MS (Patsopoulos et al., 2011). “We don’t really understand why that would be, but it’s an interesting observation,” De Jager says.
Identifying how the genes might be contributing to MS is not a trivial task. "That's the crunch facing us right now," says Trevor Kilpatrick, a neurologist at the Florey Neurosciences Institutes at the University of Melbourne in Parkville, who heads one of the two Australian IMSGC groups. But researchers are slowly starting to tie the known MS risk genes to biological processes that could affect disease onset (see "Altered Immunity, Crippled Neurons"). Kilpatrick’s group is studying a family of proteins known as the TAM receptors, one of which—MERTK—is associated with MS. Changes in signaling by these receptors, his research suggests, could affect nerve cell recovery after demyelination (Ma et al., 2011; Binder et al., 2011) and modulate autoimmunity. At least one plasma-borne TAM receptor ligand shows initial promise as a biomarker of disease, Kilpatrick said during a presentation at the November 2011 Society for Neuroscience meeting in Washington, D.C. Harbo’s group is analyzing the gene CLEC16A, which had one of the strongest associations in the most recent GWAS, and has found that the risk allele affects the production of one form of the protein it encodes in the thymus; the protein’s role there, however, is unknown (Mero et al., 2011). Such studies are beginning to show how some genes might be involved in MS, but they so far provide only the barest hints of mechanism.
Understanding the functions of genes uncovered by GWAS starts with mapping the precise location of the SNPs that identify a risk variant. This venture can be tricky. Some SNPs lie in the protein-coding section of the gene, making it likely, researchers say, that the genetic spelling variations correspond to codon differences. But the vast majority of SNPs reside in difficult-to-interpret areas, either in introns—stretches within a gene that can influence which versions of it are transcribed—or in even murkier regions between genes. Furthermore, a variant on one part of the genome can control a gene on a completely different chromosome. Often, the matchup between SNP and gene is a best guess, made by choosing the closest known gene. Sometimes the gene can be identified by a process called fine-mapping, in which researchers use an increasing number of markers to home in on sequences in nearby regions, “but a lot of these areas just won’t break down with fine-mapping,” Kilpatrick says. His group identified one risk locus on chromosome 12 that could have been tied to any one of 17 genes. Researchers ascribed it to a gene called CYP27B1, which is involved in converting vitamin D to its active form, simply because that made the most sense, he says, but that is a so-called biased approach. Indeed, his group’s subsequent fine-mapping and sequencing studies suggest they’ve got the wrong gene—and that’s not an isolated incident, he says: “You’ve got to be careful about pinning the tail on the donkey."
Critics often note that the millions spent on GWAS have brought researchers no closer to understanding MS—or any disease—or to new therapies. Progress might not be linear, however; some argue that it will come only when a critical mass of risk variants is identified. “I think the first wave of GWAS [for MS], which identified a relatively small number of genes, wasn’t all that helpful,” says Tim Behrens, senior director of immunology diagnostics and biomarkers at Genentech in South San Francisco. But with 50 or 100 SNPs associated with the disease, “we can start mapping to hopefully coherent pathways.”
Uncovering missing heritability
Each gene identified by the IMSGC represents a minuscule portion of the disease’s heritability, at least in the simplest analysis. Researchers use a measure called an odds ratio to describe the size of an effect; the greater than 1 it is, the greater the likelihood that the SNP or gene in question is associated with the disease. HLA DRB1*1501 has an odds ratio between 2 and 3, meaning that one’s likelihood of getting MS is doubled to tripled relative to the general population; odds ratios for the non-HLA genes hover around 1.1 or 1.2—that is, a 10% to 20% increase in risk. Because most people’s risk of getting MS is fairly low—a reasonable estimate is 3:1000 in the U.K. (Alonso et al., 2007) and the U.S. rate is believed to be similar—a 20% increase hardly changes it. Researchers say that the 60 genes outside the MHC together explain only a small percentage of MS’s heritability, significantly less than that explained by the MHC. IMSGC scientists posit that even with small effect sizes, genes could reveal pathways that play a central role in the disease. Others, however, have raised doubts that this is the case. “I think the experiment they embarked on was worth doing,” Ebers says. But, he adds, “if I was spending this amount of time and money on this and I came up with genes that explain [so little] of the risk, I’d be disappointed.”
Part of the problem might lie in the assumptions underlying the interpretation of GWAS data. Risk alleles are generally treated additively in statistical analyses, simply because this is the easiest way to model the enormous amounts of data that GWAS produce. With an additive approach, the more alleles you carry, the higher your theoretical risk. The method, however, does not incorporate the possibility that genes’ activities could combine in more complicated ways. In May 2011, Michael Demetriou, an immunologist at the University of California, Irvine, identified a connection between MS and disruption of a process called N-glycosylation, which attaches complex sugars to proteins and can modify their function (Mkhikian et al., 2011). The IL2RA, IL7RA, and MGAT1 risk alleles—but not those associated with other genes—disrupt N-glycosylation and cause symptoms characteristic of MS in mice. When the three are present together, however, MGAT1 negates the deleterious effects of the other two. Vitamin D delivers the same mitigating effect, revealing a complex interplay of genetic and environmental factors. “If some of these genes have an odds ratio of only 1.2, I don’t think it’s because they contribute such little risk,” Demetriou says. "It’s just that there are these other factors canceling them out.”
Demetriou and others have proposed that such nonlinear combinations might explain why the known crop of genes seems to identify only a portion of the disease’s heritability; if examined correctly, the genes might explain much more of it. A paper published in January by Lander’s group at the Broad Institute in Cambridge, Massachusetts, suggests that this idea might be generally true for illnesses associated with relatively common gene variants, but identifying such interactions might require enormous studies of hundreds of thousands of patients (Zuk et al., 2012). “This is a monumental task,” Baranzini says, adding that researchers have begun examining pairs of genes to assess whether disease risk increases more than additively when both are present. So far, however, no clear method exists for systematically detecting how risk variants enhance or slash one another’s effects, he adds. Results to date suggest that common variants do appear to behave additively, De Jager says, “but the problem is that the studies are underpowered to detect” synergistic interactions that might be present. Gene interactions might be particularly relevant in specific subsets of patients, he says, but currently no meaningful approach parses out such groups. Hillert’s lab is working on a database that will tie information about individuals’ disease course and relevant environmental factors—whether they smoked or were infected with Epstein-Barr virus (EBV), for example—to their genotype in order to begin systematically searching for such interactions (see "Viral Villain").
Researchers within and outside the IMSGC propose several other possibilities for where the missing heritability might lie. First, with GWAS to date covering only about 80% of the genome, additional common risk variants might lie in areas with poor SNP coverage. Additionally, Baranzini notes, risk alleles might be distributed along key pathways that are misregulated in the disease. For example, a particular SNP associated with the disease might be found in, say, 20% of the population. But another SNP, present in another 20% of the population, might participate in the same physiological process as the first one. Uniting all of these small effects under the umbrella of a single pathway might flush out more of the missing heritability.
Another avenue of exploration is rare variants that occur sporadically in certain families but are uncommon in MS patients in general. Such variants can’t easily be found with GWAS; rather, their identification requires whole-genome sequencing or at least sequencing of the coding region of the genome and comparing the results among affected and unaffected family members. Techniques to carry out such experiments are becoming available, and Baranzini and his colleagues have sequenced DNA from 12 individuals in one family: siblings, cousins, and an aunt, of whom six have the disease and six do not. The researchers are now combing the genomes for variants associated with the disease. “I’m not 100% sure we’re going to find something, but I’m 100% sure we should try,” he says.
Still another possibility is epigenetics: changes in gene activity caused not by differences in the gene’s DNA sequence but by processes that affect how the DNA is read. Examples include methylation of DNA; acetylation of proteins called histones, around which chromosomes wind; and N-glycosylation, the process that Demetriou studies. A handful of reports have suggested that the risk of developing MS differs depending on whether individuals inherit a particular allele from their father or their mother. Such a pattern would arise if the chromosomal regions harboring these genes are “imprinted,” carrying epigenetic marks that turn genes on and off.
Meanwhile, IMSGC researchers are focusing on the identified risk alleles to determine whether they can somehow be used to predict the chances that an individual will get MS—not in the general population, but in people who have a close relative with it or who have experienced an isolated neurological episode or have MS-like lesions. De Jager’s group is developing an algorithm that uses genetic and environmental information to stratify such people based on their risk. The researchers are now conducting a study to determine which, if any, of these factors correlate with asymptomatic disease, as captured by MRI. “This population is one in which you might consider intervening in some way,” he says—for example, giving them vitamin D as a preventive measure, or monitoring them more closely. Down the line, the payoff may be bigger still. “We’re nowhere close to doing this,” De Jager says, “but we may one day be able to create some sort of vaccine” for the disease.
Although the MS genetics field has inched along until the last 6 months, the recent advances have brought its work to a turning point, De Jager and others say. With the identification of the common risk variants for the disease close to its logical conclusion, researchers are now well positioned to transform current knowledge into a springboard for the next stage of research.
Despite the complexity and the caveats, IMSGC researchers say, the investment so far has been well worth it. “If you’re an optimist, we have succeeded beyond expectations,” Hafler says. “If you’re a pessimist, my response is, 'Come on, we’re just starting.' ”
Correction (20 April 2012)
The story has been corrected to clarify the origin of the IMSGC.
Key open questions
- What clinical or other factors could be combined with genetics to predict risk in individuals who have family members with MS?
- How can novel statistical and computational methods help identify possible gene interactions that raise the risk of getting the disease?
- How can researchers effectively identify changes in gene expression or epigenetic factors and their role in disease risk?
- How much do rare variants explain the missing heritability of the disease, if at all? How much do they uncover novel disease mechanisms?
- What can genes that are associated with other autoimmune disorders contribute to researchers’ understanding of MS?
- Are any genetic markers associated with subpopulations of patients (according to disease type or lifestyle factors, for instance) or disease factors such as viral status or response to specific therapies? Initial studies don’t show such a breakdown, but researchers say they haven’t yet established a good approach for addressing this question.
- What experiments would resolve the debate about whether the immune system or the nervous system is the first source of pathogenesis in MS?
Image credits
Thumbnail on landing page. "A DNA Microarray," Guillaume Paumier/Wikimedia Commons, 2008. Released under a Creative Commons Attribution-ShareAlike 3.0 Unported CC BY-SA 3.0 license.
Fig. 1. Adapted with permission from Macmillan Publishers Ltd: Nature Reviews Genetics. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. JR Oksenberg, SE Baranzini, S Sawcer, SL Hauser, 9: 516-526, copyright 2008.
Fig. 2. Female/Male Shadows, Mikael Häggström/Wikimedia Commons, 2009. Released into the public domain.
Fig. 3. Courtesy of Alla Katsnelson.
Suggested by philip de jager
This paper by the International MS Genetics Consortium (IMSGC) is an important milestone, as it reported the discovery of common genetic variants of modest effect that are associated with MS susceptibility using a whole genome scan approach. It therefore provided robust evidence that (1) susceptibility alleles of this type exist outside of the MHC and (2) a genome scan strategy was appropriate for discovering such variants. It has therefore been the foundation for subsequent studies by the IMSGC and other groups of investigators that have gradually elaborated the genetic architecture of MS susceptibility. The model that the genetic component of MS susceptibility results from the contribution of risk alleles in many different genes is now well accepted. This study also provided an important illustration of the power of consortia of investigators in successfully exploring challenging questions in MS.