Neuromyelitis Optica, Part 1: The Other Disease
Once thought of as a variant of multiple sclerosis, neuromyelitis optica is now clearly understood to be a different disease
Lea Long still remembers the moment her life changed like it was yesterday. She was in the supermarket standing in line to pay one day in 1998, when she all of a sudden felt as though she was going blind in her left eye. “[It] was like a window shade coming down,” she said in an interview with MSDF. Long called the babysitter who was with her three small children to take her to the hospital. There, doctors told her that she had inflammation of the optic nerve. To tame the inflammation, they gave her steroids. The next day, she had lost all sight in her left eye, but after two or three months, it came back.
In 1999, she had another attack. This time, most of her body went numb; she couldn’t walk and couldn’t even use a fork. “I lost the use of [my] legs, arms, everything,” she said. “[I] couldn’t feed myself.” Again, doctors gave her steroids. After dozens of tests came back negative, they concluded that she had multiple sclerosis (MS). They prescribed weekly intramuscular injections of interferon beta 1a (Avonex), a drug commonly used to treat MS.
This time, recovery took much longer. Because Long couldn’t type, drive, or dial a phone, she had to quit her job as a business development manager for a high-tech company. During 6 months of physical therapy, movements slowly came back, and after a year, she could finally run again.
In the following years, she had additional relapses, and in 2009, she lost the sight in her left eye again, this time for good. After that episode, Long finally learned that she really had a different demyelinating disease called neuromyelitis optica (NMO), and that the MS drug she had been taking all those years wasn’t the right treatment. In fact, in some cases it can make NMO worse.
Long isn’t the only NMO patient who has been misdiagnosed. NMO is much less common than MS in the U.S., and sometimes doctors mistake it for MS, because the symptoms are strikingly similar: Both disorders often impair vision and walking, and both involve abrupt attacks when symptoms worsen that are punctuated by periods without any attacks. What’s more, both show an onset around 30 or 40 years of age, and both are more common in women.
Because of these similarities, doctors assumed until a few years ago that NMO is a variant of MS. It was originally known as Devic’s disease, because in the late 19th century, French neurologist Eugene Devic was the first clinician who reviewed published cases of patients with simultaneous optic neuritis (inflammation of the optic nerve) and myelitis (inflammation of the spinal cord).
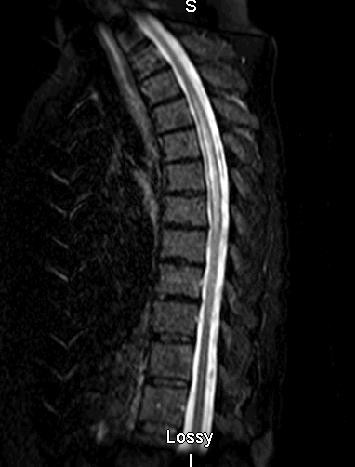 But subtle differences distinguish NMO from MS: NMO attacks are generally more severe and sometimes fatal. Both diseases involve central nervous system (CNS) lesions, which can be seen by MRI, but in NMO patients, these lesions are typically found on the spinal cord (often affecting three or more vertebral segments) and optic nerves, whereas MS often affects other brain areas. And while MS is more common in temperate zones of both hemispheres, NMO seems to equally affect individuals from different ethnic groups and climates. That’s why, among people from the West Indies and Asia, NMO appears to be a quite common cause of demyelinating disease, while it is far less common than MS among white people in North America, Europe, or Australia. (In the U.S., only about 4,000 to 10,000 people have been diagnosed with NMO, compared with an estimated 400,000 with MS.)
But subtle differences distinguish NMO from MS: NMO attacks are generally more severe and sometimes fatal. Both diseases involve central nervous system (CNS) lesions, which can be seen by MRI, but in NMO patients, these lesions are typically found on the spinal cord (often affecting three or more vertebral segments) and optic nerves, whereas MS often affects other brain areas. And while MS is more common in temperate zones of both hemispheres, NMO seems to equally affect individuals from different ethnic groups and climates. That’s why, among people from the West Indies and Asia, NMO appears to be a quite common cause of demyelinating disease, while it is far less common than MS among white people in North America, Europe, or Australia. (In the U.S., only about 4,000 to 10,000 people have been diagnosed with NMO, compared with an estimated 400,000 with MS.)
And while the cause of MS is still largely unknown, in 2005 researchers found a biomarker—and likely cause—for most cases of NMO: an antibody that attacks a molecule called aquaporin 4 in the optic nerve and the spinal cord. This discovery enabled them to develop a diagnostic test for aquaporin-4 antibodies in the blood. The test, available since late 2005, enabled doctors to finally diagnose Long’s NMO.
The aquaporin-4-antibody finding has also enabled researchers to define a broader spectrum of NMO-related disorders than was previously known, to study how NMO develops, and to find better drugs to treat and prevent NMO attacks, some of which are now in early clinical trials. Since the antibody discovery, “It’s been a dramatic exponential rise in publications [on NMO], from a few a year to thousands,” said Sean Pittock, M.D., in an interview with MSDF. Pittock is an autoimmune neurologist at the Mayo Clinic in Rochester, Minnesota, and was involved in the discovery.
These insights about NMO may even hold a lesson for a better understanding of MS: Perhaps, some researchers say, the 2.1 million people with MS worldwide harbor other disorders that are really caused by antibodies. Evidence for this is already emerging.
A landmark discovery
The causes of MS are still unclear: Researchers have been unable to find a clear culprit, such as reproducibly confirmed autoantibodies—antibodies against targets in the body’s own tissues like the CNS—that could explain the symptoms. For a long time, they assumed that this was also the situation for NMO, which they thought was a subtype of MS.
That thinking started to change in the late 1990s, when researchers noticed that many NMO patients do, in fact, have autoantibodies. Brian Weinshenker, M.D., a neurologist from the Mayo Clinic in Rochester, Minnesota, found that people with NMO carried antibodies to cellular proteins such as those found in the cell’s nucleus and mitochondria, the cell’s “power plants.” When he gave a talk about his findings in 1998, his Mayo colleague Vanda Lennon, M.D., Ph.D., was intrigued. “I said to him after the talk, ‘This is certainly not MS,’ because I [had] never seen any antibodies of note in MS [after 3] decades of looking,” Lennon told MSDF.
Indeed, Lennon had a lot of experience looking at antibodies: At the time, she was running not only her research lab but also a diagnostic lab that tested whether patient sera contained antibodies that bind to certain parts of the CNS in sections of the mouse brain. Some of these autoantibodies can cause neurological symptoms; often, they are induced by CNS proteins made by cancers of the breast, ovary, thymus, or lung. To this day, Lennon’s lab has been analyzing samples from tens of thousands of patients from all over the U.S. every year.
Lennon asked Weinshenker for serum samples from his NMO patients. When she checked whether they contained antibodies that bind to parts of the mouse brain, she noticed a unique pattern of staining on the outside rim of blood vessels.
She remembered that she had seen the same pattern before—in 12 samples from the reference lab that didn’t fit any known disease. When she checked those patients’ clinical histories, she was intrigued to find that they all had NMO-like symptoms: inflammation and spinal cord damage. “That’s when we knew we were really onto something,” Lennon said. The final piece of the puzzle fell into place when additional samples from 102 NMO patients showed the same pattern. Lennon eventually published the finding in The Lancet (Lennon et al., 2004).
Still, what was the antibody targeting? It took Lennon only a year to find out: She realized that the distribution pattern of the antibody targets included tissues outside the CNS such as the kidney. This is similar to the distribution of a protein called aquaporin 4, which is found on the surface of the brain’s astrocytes as well as in the kidney. In 2005, Lennon reported that aquaporin 4 was indeed the target of the antibody in NMO patients (Lennon et al., 2005).
“That totally defined the disease,” Alan Verkman, M.D., Ph.D., told MSDF. Verkman, a clinician-scientist at the University of California, San Francisco, was involved in the study.
Two different diseases
The availability of the aquaporin-4 antibody test helped physicians to correctly diagnose NMO and distinguish it from MS. This task is difficult, especially if patients visit the doctor outside of an acute attack and MRI results are unclear. But a correct diagnosis is important, because some of the drugs doctors use to treat MS—such as the interferon beta drug Long was initially treated with—can make NMO worse.
Before the test was available, there was often no way to distinguish MS from NMO, said Angela Vincent, MBBS, a neuroimmunologist at the University of Oxford, U.K. “Although some people thought they were two distinct diseases,” she told MSDF in an interview, “others weren’t convinced, and some of the patients are very, very similar clinically, at least when you first see them.” And even though the test has now been available for years, a certain percentage of patients still thought to have MS may very well have NMO, said Mayo researcher Claudia Lucchinetti, M.D., in an interview with MSDF.
NMO was originally considered a disease of the optic nerve and spinal cord, with limited involvement of the brain. But once the aquaporin 4 test became available, researchers and doctors realized that NMO can involve a wider spectrum of disorders called NMO spectrum disorders, such as inflammation of just the spinal cord or the optic nerve, narcolepsy, or intractable nausea.
Pittock recently reported that as many as 14% of NMO patients initially suffer from intractable nausea (Iorio et al., 2013). Doctors don’t usually think of NMO or MS when they see such patients, Pittock said, which means that it can take a long time until patients receive the correct diagnosis. Recently, Pittock saw a 23-year-old African-American patient who woke up one day, felt nauseated, vomited for 6 months, and lost 25 pounds as a result. The patient consulted countless gastroenterologists, Pittock said, none of whom made the correct diagnosis. Then the vomiting stopped, and a few weeks later he developed problems walking, which is when he finally saw Pittock and was diagnosed with NMO.
____________________
Continue to Part 2: Pathogenesis