Genome-wide Study Brings MS Landscape Into Sharper Focus
New study increases to 110 the number of genes closely linked to MS
The International Multiple Sclerosis Genetics Consortium (IMSGC) has announced the discovery of 48 additional variants in the human genome that contribute to susceptibility to MS, catapulting the scientific community closer to identifying the genetic architecture underpinning susceptibility to the disease (IMSGC, 2013).
“The new findings double the information we have on unraveling the genetic and environmental elements involved in susceptibility to MS, while adding more potential therapeutic targets,” Fred Lublin, M.D., director of the Corinne Goldsmith Dickinson Center for Multiple Sclerosis at the Icahn School of Medicine at Mount Sinai, said in an interview with MSDF.
The 48 new variants, together with others the consortium published in 2011 (IMSGC, 2011), total 110 sites in the human genome that are part of or closely linked to genes that contribute to inherited risk of developing MS. The new results also indicate that it isn’t simply identifying the genes that will prove most informative but understanding how they interact. “These new results underscore the fact that pathways and networks of interactions between genes are likely to be more important than individual genes,” said team member Stephen Sawcer, M.D., Ph.D., a clinical neuroscientist at the University of Cambridge, U.K., in an interview with MSDF.
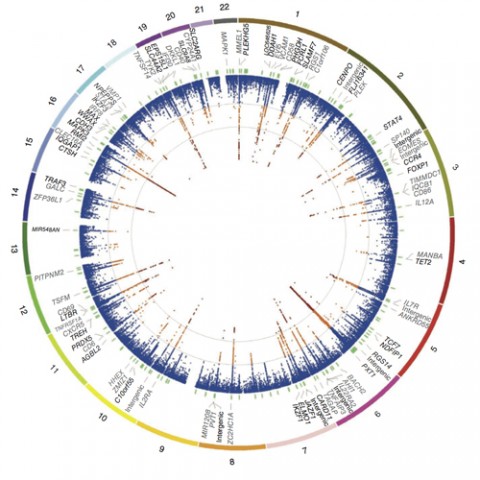
The role of genetics in MS is complex in the technical sense of the word; many genes interact with each other and with environmental factors to set the disease in motion (see Genetic Associations). But different genes, and their variants, don’t contribute equally to the disease process, and their input may vary among individuals. For these reasons, there is no simple answer to the seemingly simple question, “I have MS; will my child develop it too?”
In addition to estimating risk, genetic information can offer clues to how symptoms arise and evolve. “Every gene we identify is like a piece in an enormously complex jigsaw puzzle. If you have only one piece, it’s hard to work out what the picture is. If you have two pieces, that’s slightly better. But if you have 100 pieces, you can start to fit the pieces together and to recognize patterns. The more puzzle pieces you have, the more likely they are to reveal clues about how and why MS develops,” said Sawcer.
From twins to genome-wide sweeps
MS has long been known to cluster, if not “run,” in families; it’s more common among relatives of individuals with the disease than in the general population. In the pregenome era, studies of twins, adopted individuals, and other special categories of relatives led geneticists to conclude that this clustering was mainly due to the influence of genes rather than environmental factors (Hawkes and Macgregor, 2009).
Yet studies of people who migrated from one country to another suggested that environmental factors, such as diet and exposure to pathogens, could account for some of the differences in frequency of MS in different parts of the world. “Twin studies over the years always led us to understand that there is a strong genetic component, but they clearly don’t explain everything,” Jacob McCauley, Ph.D., lead investigator in the current study and a genome researcher at the University of Miami Miller School of Medicine, told MSDF.
While twin and adoptee studies estimate the relative size of the genetic component of a trait, genomic tools point to specific genes. Genome-wide association studies (GWAS), which the consortium uses, grew out of the results of the Human Genome Project. These investigations catalog single-nucleotide polymorphisms (SNPs)—single-base sites that differ in frequency in populations.
Using a GWAS, researchers can test many thousands of different SNPs at the same time, in many people, to discover whether a specific SNP is significantly more common among people who share a trait or illness compared to controls. A GWAS reveals a number of genome regions that each contributes a very small amount to the variability of the complex trait or illness.
A directed search
The new study evaluated 161,311 SNPs among 14,498 people with MS and 24,091 controls. The investigators then tested the 135 most strongly associated SNPs in an additional 14,802 cases and 26,703 controls. The project therefore involved more than 80,000 individuals and in total identified 48 new susceptibility gene variants. Combined with already known genes, such as two interleukin receptor genes (IL2RA and IL7RA) and variants within the HLA-DRA locus of the major histocompatibility complex (MHC) (IMSGC, 2007), the new work brings the total to 110 risk variants at 103 sites in the genome.
The researchers used the ImmunoChip from Illumina Inc. A DNA “chip” or microarray is a solid grid that holds pieces of selected genes immobilized at specific positions. A researcher applies labeled DNA samples, and the resulting signals emanate from SNPs that have found and bound their complements on the grid. The ImmunoChip paints a portrait of a person’s variability for the displayed genes that affect immunity.
“Groups for different autoimmune diseases—type 1 diabetes, inflammatory bowel disease [IBD], lupus, celiac disease, and others—came together and asked, ‘Why don’t we create a cost-effective way to do lots of samples?’ ” McCauley recalls. The chip holds DNA pieces from 184 parts of the genome outside the MHC where variant SNPs mark at least one autoimmune disease. Twenty-two percent of MS-associated SNPs are seen in one or more other autoimmune diseases, including IBD, primary biliary cirrhosis, rheumatoid arthritis, psoriasis, and autoimmune thyroid disease.
“The idea of generating an ImmunoChip to selectively study non-MHC loci with genome-wide associations to at least one autoimmune disease is very clever, since it allowed the researchers to define new susceptibility variants and refine the overlap between MS susceptibility and other autoimmune diseases,” said Patrizia Casaccia, M.D., Ph.D., a neurogeneticist at Mount Sinai, in an interview with MSDF.
SNPs with very strong associations may highlight genes directly implicated in the pathogenesis, which may reveal targets for drug discovery or repurposing. For example, one SNP is in a gene called SLC44A2 that regulates another gene, ILF3, which affects T cell expression of interleukin-2, controlling inflammation.
Although a GWAS generates more precise data than a twin study, it typically doesn’t explain all the observed clustering in families. In the new study, the 110 risk variants explain 20% of the sibling recurrence risk, which increases to 28% when including the MHC effects. “While it might be tempting to see the ‘missing’ 72% as failure of the approach, it might also be viewed as an opportunity to find out yet more about the disease,” Sawcer explained.
An immunological footprint
Deploying a DNA chip loaded with SNPs from immune-related genes may be stacking the deck toward discovering immune-related gene variants, but targeting the search makes economic sense. “There’s a lot to be learned and shared among individual diseases. We’re trying to understand how genes come together, and how their expression turns on and off in MS,” said McCauley.
And many of the genes interact. “The 110 variants are overwhelmingly related to regulating the balance of expression of different genes. We do not understand that language,” said Sawcer.
The new results favor the inflammation hypothesis but aren’t the final word. “The genetic data confirm the age-old contention that MS is primarily inflammatory. Until now that view has largely been based on circumstantial evidence, but now we have truly objective evidence,” Sawcer said.
Casaccia agreed that the ImmunoChip results support the role of autoimmunity in the relapsing-remitting form of the disease, but added, “It would be interesting to design similar approaches that interrogate gene susceptibility networks related to neurodegenerative disorders and run similar studies for patients with the primary progressive form of the disease, where the pathogenic mechanism cannot be explained by the autoimmune hypothesis.”
Key open questions
- What accounts for the “missing clustering” of MS in families?
- How many more SNPs must be added to GWAS to saturate the genome?
- What environmental factors affect susceptibility to MS?
- How can we identify gene-gene and gene-environment interactions that contribute to MS susceptibility?
- Which genes that are not involved in immunity contribute to MS?
- What are clinical applications of the GWAS results?
Disclosures
Dr. Lublin acknowledged receiving funding from Acorda Therapeutics, Biogen Idec, Novartis, Teva Neuroscience, Genzyme, Sanofi, Celgene, the NIH, and the NMSS; serving as a consultant or advisory board member for Bayer HealthCare, Biogen Idec, EMD Serono, Novartis, Teva Neuroscience, Actelion, Sanofi-Aventis, Acorda, Questcor, Roche, Genentech, Celgene, Johnson & Johnson, Revalesio, Coronado Bioscience, Genzyme, MedImmune, Bristol-Myers Squibb, Xenoport, Receptos, and Forward Pharma; and owning stock in Cognition Pharmaceuticals. Drs. Casaccia, McCauley, and Sawcer reported no relevant financial interests.
This paper by the International MS Genetics Consortium (IMSGC) is an important milestone, as it reported the discovery of common genetic variants of modest effect that are associated with MS susceptibility using a whole genome scan approach. It therefore provided robust evidence that (1) susceptibility alleles of this type exist outside of the MHC and (2) a genome scan strategy was appropriate for discovering such variants. It has therefore been the foundation for subsequent studies by the IMSGC and other groups of investigators that have gradually elaborated the genetic architecture of MS susceptibility. The model that the genetic component of MS susceptibility results from the contribution of risk alleles in many different genes is now well accepted. This study also provided an important illustration of the power of consortia of investigators in successfully exploring challenging questions in MS.
Comments
MS twin studies, although widely cited to infer a “30% genetic contribution,” are uninterpretable principally because of small numbers and large confidence intervals as detailed earlier (Hawkes and McGregor, 2009). Despite this, the search for mutations that might be the root cause of MS goes on and on.
Using the latest Illumina technology, not a single mutation has been found, yet over a similar time period utilizing similar gene-crunching methods, numerous mutations have been discovered in familial parkinsonism, peripheral neuropathy, spinal muscular atrophy, and the muscular dystrophies, to name but a few. Normally such persistently negative findings in MS would deter further genetic investigation, but no, we now look for normal variants (polymorphisms) or changes in DNA that make us tall or short, blue-eyed or brown-eyed, etc. Most diseases have a set of polymorphisms that confer susceptibility; e.g., those with blood group O are more vulnerable to peptic ulcers (Clarke, 1959), but the basic cause is now known to be an infection.
It is now proposed that there may be gene-gene interaction (epistasis) or even modification of DNA expression by environmental agents (epigenetics). A far more compelling answer is that MS has an unimportant genetic component and that it is due to an infection—possibly one that cannot be disposed of once it enters the brain such as the Epstein-Barr virus—but there are other contenders such as exogenous or endogenous retroviruses. It is now time to concentrate more on the possibility of an environmental cause for MS. MS genetics has probably had its day (Hawkes, 2013).
Christopher Hawkes, M.D., Barts and the London School of Medicine and Dentistry, London
Clarke CA. Correlations of ABO Blood Groups with Peptic Ulcer, Cancer, and Other Diseases. Am J Hum Genet. 1959 June; 11(2 Pt 2): 400–404. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1932162/pdf/ajhg00571-0114.pdf
Hawkes CH. Multiple sclerosis genetics is dead. Multiple Sclerosis and Related Disorders. Volume 2, Issue 3, July 2013, Pages 156-161. http://www.msard-journal.com/article/S2211-0348(12)00155-1/abstract