MS Genetics: The Outer Limits
In a mashup of data sets and new genome-mapping tools, a study finds that most of the inherited risk for autoimmune disease discovered so far comes from mysterious noncoding regions and mostly affects immune cells
Since 2007, ever larger and more powerful genome-wide association studies (GWAS) have found over 150 genetic variants that are more common in people with multiple sclerosis (MS), compared to those who do not have the disabling inflammatory disease of the brain and spinal cord. But there’s a catch.
That impressive number refers mostly to signposts marking chunks of chromosomes where researchers have assumed a guilty gene lurks. The risk from each variant is low, but in an unlucky mix they can add up. The reason for doing GWAS hasn’t changed: Find the specific genetic variants, explore the contributing molecular pathways, and develop better therapies tailored to the disease.
But an uncomfortable question has crept into the discussion about exactly how to pinpoint the culprits.
“What if we find them, and they’re not in the genes?” That’s one of the questions Alexander Marson, M.D., Ph.D., of the University of California, San Francisco, and his colleagues asked 5 years ago when they launched their study. It doesn’t take a computational biologist to figure those odds. Protein-coding genes make up about 2% of the human genome. Although researchers have only begun to probe the secrets of the vast noncoding regions, their mysterious functions seem to be important in health.
Last week, the team’s findings confirmed what many had begun to suspect. Nearly 90% of the slightly risky variants known so far for MS and other autoimmune diseases lie outside of protein-coding genes. About 60% fall on DNA addresses known as enhancers or switches, which manage gene activity in mostly enigmatic ways.
The study, published online October 29 in Nature (Farh et al., 2014), contributes a trio of research tools, developed with MS data and generalized to other diseases, said Marson, a co-first author. First, it presents a new statistical way to more finely map disease variants from high-powered GWAS with more certainty.
Second, the team created maps of the DNA regions open for enhancer activity in various types of stimulated immune cells, filling in gaps in the epigenomic data collected for human cells.
Finally, when they combined the overall genetic risk maps with the distinctive enhancer maps of immune and other cells, the researchers picked out the cells most likely at the heart of MS and other autoimmune diseases.
A person’s genome may be the same throughout the body, but a neuron deploys different genes than a skin cell. Other genes are locked down in the DNA’s chromatin packaging. Each gene used by a cell, in turn, has an entourage of enhancers at other places in the genome, sometimes far away, that help control the gene in different ways. Some enhancers are well-known parking places for transcription factors that can boost or repress gene activity. But, as this study points out, most of the likely disease variants fall beyond the limits of scientific knowledge.
“It took us a long time to accept that,” Marson told MSDF.
The paper provides a framework for identifying functional variants outside of protein-coding genes. It also frames a new challenge in understanding what goes wrong in disease. Scientists can read a DNA sequence to know what protein a gene makes and then test how a disease-associated version of the gene works, but they haven’t cracked the complex regulatory code that turns a gene on and off at the right time and tells it how much protein or RNA to make.
“These mutations are sitting on switches with subtle effects on gene regulation,” said co-author Bradley Bernstein, M.D., Ph.D., of the Broad Institute of MIT and Harvard and Massachusetts General Hospital. “It’s not immediately obvious how they are changing the switch. What they would do is not explained by current regulatory models. We have a lot of work to do.”
On the other hand, Bernstein pointed out, the findings suggest a possible therapeutic direction. Even if scientists don’t completely understand how they work, experimental epigenetic drugs with possible immunomodulatory effects have been developed and are being tested in clinical trials for cancer, he told MSDF.
View of the MS genome
The study adds more evidence that MS begins as an autoimmune disease outside of the central nervous system, said co-author David Hafler, M.D., of Yale University in New Haven, Connecticut, a point he hammered home in his keynote talk at the recent joint American-European MS meeting in Boston. More importantly, he told MSDF, “This is a road map to study MS. It moves the genetics to the next level. We hope other biologists will jump on this data.”
Understanding the combinations of risk genes in individuals with MS may eventually lead to more tailored treatments, but that’s down the road. “We’re looking at variants that dictate risk of developing MS,” Hafler said. “We’re not looking at the phenotype of what leads to disease progression. Some people have more inflammation and recover. Some have less inflammation and do badly. As we map genetics of that phenomenon, we will see variants in the nervous system.”
The new study sought to map common inherited variants, but some risky nicks and dings to DNA’s chromatin packaging may happen after birth. MS is widely believed to arise from a combination of inherited and environmental factors. Smoking, infections, and low vitamin D have been implicated as triggers or additional susceptibility factors. Some may interact badly with the causal variants (Briggs et al., 2014), or they also may exert their influence by tweaking the chromatin.
The study by Marson and his colleagues “does not directly address the role of environmental stimuli on genomic loci,” Patrizia Casaccia, M.D., Ph.D., at the Icahn School of Medicine at Mount Sinai in New York City, wrote in an email to MSDF. “The chromatin landscape is highly cell specific and the changes occurring in immune cells are not predictive of those occurring in neurons or oligodendrocytes in the brain.”
Her lab, for example, has shown that the seemingly unaffected areas in brains of people with MS harbor epigenetic changes associated with decreased expression of protective genes and may be more susceptible to damage (Huynh et al., 2013).
“It is conceivable that the environment might have a dual effect: On the immune cells, it may contribute to open chromatin confirmation in regions associated to active enhancers of immune-related genes, while in the brain it may favor chromatin conformations or changes in DNA methylation that modulate the responsiveness of cells to damaging stimuli,” Casaccia wrote.
“It would be of high interest to define whether the distinct associations and modalities of regulation occur in primary progressive MS patients,” she added. The MS GWAS studies underlying this study were conducted by the International Multiple Sclerosis Genetics Consortium (IMSGC), which Hafler co-founded. The group is turning its attention to progressive forms of MS, but so far the genetic data mostly includes people with relapsing-remitting MS, the most common form.
Algorithm fine mapping
Thousands of people are needed for genome-wide association studies, because each variant contributes such a small risk for disease. Consortium members estimate they have found about half the inherited risk factors that explain how MS can run in families. Another limiting factor for genome-wide studies is the cost of genotyping such large numbers of people.
The discovery 10 years ago that DNA is passed from parents to children in chunks called haplotypes allowed researchers to sample the genome less expensively by using telltale markers in the DNA sequence, called single nucleotide polymorphisms (SNPs). The haplotype concept and methods, developed in part by Mark Daly, Ph.D., of the Broad Institute in Cambridge, Massachusetts, a co-author of the new study, allowed for a new era of scientifically sound and efficient GWAS, but it still leaves a gap between the genes and the biology.
About 5 years ago, a group of scientists leveraged the idea that immune-mediated diseases may be more similar than different. They developed the Immunochip, a cost-effective way to narrow the search for common causative variants to about 200,000 SNPs in 186 DNA regions with risk factors shared by multiple immune-mediated diseases.
Like a pair of binoculars, the Immunochip dials in more details, filling in more potential disease-causing SNPs on key haplotypes. “If you see a neighboring SNP in a locus, it can be 10 times more likely to be causal,” Marson told an audience in July at the Federation of Clinical Immunology Societies (FOCIS) meeting in Chicago.
For one part of the study, Marson teamed up with Broad colleagues Kyle Kai-How Farh, M.D., Ph.D., in Daly’s lab to develop a new statistical way to better estimate the causal genes. “If you look at the history of human genetics, you can appreciate that the genome is really large,” Farh told MSDF. “As we have tried to figure out the actual mutation that is causing disease, we have gone from mapping the disease to the whole chromosome, to the region, and most recently to the vicinity of the gene. We haven’t been able to figure out the exact mutation.”
They developed an algorithm using the more detailed data from the first published Immunochip GWAS for MS (IMSGC, 2013). They extended the method to seven different diseases, based on data from the 1000 Genomes Project, and generalized it for a wide range of conditions, including 21 autoimmune diseases and 18 other conditions. The team was able to identify causal variants with high confidence for about one-third of the GWAS hits.
The algorithm, named Probabilistic Identification of Causal SNPs (PICS), estimates the chances that an individual SNP is a causal variant by leveraging what is known about the haplotype, the pattern of SNP associations, and small genetic effect. Essentially, PICS runs mini-experiments on each SNP to test it as a causal variant and compares its results to the GWAS results for “the best possible guess at what the causal variant is likely to be,” Farh said.
A potential disease gene locus can have hundreds of thousands of nucleotides and contain other genes. “We’re able to make a sound statistical argument to map complex human diseases to actual nucleotide changes,” Farh told MSDF. The algorithm was used in a July paper on schizophrenia, which happened to be published before the method was formally presented in the new study (Schizophrenia Working Group et al., 2014).
Other researchers can find the candidate causal SNPs and the PICS algorithm online at the Broad. “You just plug in the numbers and go,” Farh said. But first, “you need the right genetic data and you need the right epigenetic data,” Bernstein added. “To go deep, you need to live and breathe it for a while.”
Epigenetic fine mapping
In the other main part of the study, Marson wanted to know what cells and what functions are being altered by the causal variants. For this, Marson teamed up with Bernstein to generate a new epigenetic map of specialized immune cells, pulling in data sets for other cell types.
The researchers were interested in the epigenetic patterns that reflect the way a genome is customized for the wildly different tasks required for each cell type. The team collected and mapped well-characterized immune cells from healthy donor blood, relying primarily on a histone tag (H3K27ac) linked to areas of enhancers and promoters of genes. The tag is associated with open chromatin packaging, allowing the DNA to be transcribed.
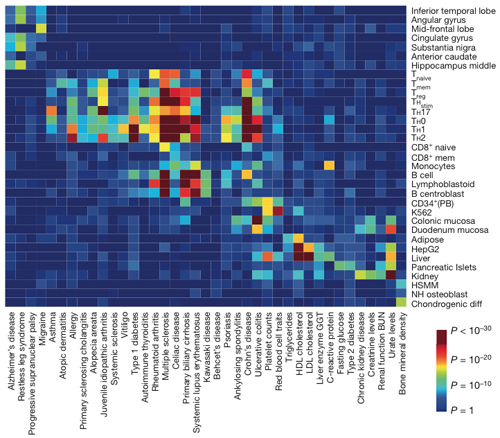
The team layered the algorithmic fine mapping over the epigenetic signatures of 56 cell types, including brain, gut, fat, liver, and bone. The causal variants for MS coincided with the cell-specific regulatory regions in immune cells. In contrast, the causal variants in Alzheimer’s disease and migraines mapped to brain tissue. By cell type, MS clustered with other autoimmune diseases, but had a distinctive signature, with the risky variants concentrated in the enhancer regions of stimulated CD4 T cells and B cells.
“My hypothesis is that the SNPs are tuning the threshold for how T cells respond to stimulation,” Marson told MSDF. In their mapping, the team mimicked how T cells respond to seeing a foreign antigen. When the T cells engage with their antigen, the team found, they turn on the set of enhancers that map to the likely disease-causing SNPs.
“It’s a whole new way to characterize diseases based on cell types with underlying genetic variants,” Marson said. “Multiple cell types might use the same gene, but might be turned on by different enhancers, so we might get more specific information about the underlying cause by looking at the enhancers.”
Many of the findings stumped the team at first. Less than half of the more finely mapped causal SNPs for MS and other autoimmune diseases landed in recognizable enhancer-binding sites. Marson and his co-authors spent a long time wrestling with the findings, thinking they were missing something.
“We were entrenched in this logic that we had to find the transcription factor motif affected by each one of these SNPs,” he said. They finally recognized that the disease variants were falling in uncharted DNA territory.
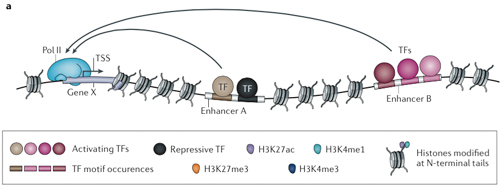
“Enhancers can be very, very far from the genes they regulate,” Marson explained. “They loop around in three dimensions. That’s why figuring out what enhancers are was hard until a few years ago.”
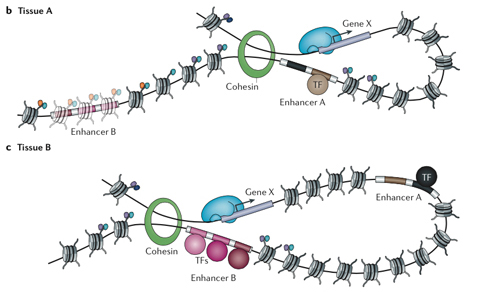
The regions between genes, formerly known as junk DNA, contain “hundreds of thousands, if not millions, of enhancers” that shape gene expression, Marson said. Some of them are repressed by the chromatin packaging of DNA and some are poised to act upon certain signals, such as a T cell gearing up to fight its viral antigen.
Some clues to the function of the likely disease-causing SNPs are emerging from studies of model organisms, such as fruit flies, Bernstein told MSDF. “The effects are subtle and not places you would predict, such as in evolutionary conserved motifs,” he said. “The flanking motifs turn out to be important.”
Next steps
Meanwhile, Hafler’s lab has followed up with experiments looking at how the genetic variants in the binding sites for one enhancer, the well-known transcription factor NF-κb, change the biology of immune cells in MS and two other autoimmune diseases in which the variants play a role. NF-κb has been implicated in previous studies as well.
Marson is following up by systematically evaluating how each risky autoimmune SNP alters each cell’s function using the new CRISPR technology to precisely edit each change into healthy cells in culture. The work is part of a new consortium, known as the Innovative Genomics Initiative.
The main idea of genome studies is to push them to find out what’s going wrong and to figure out better ways to treat it. “That’s the whole thing with genomics,” Marson said. “There’s no shortage of data. The challenge is, what can we learn from it?”
Key open questions
- How are the MS causal variants affecting enhancer regions, and what role does that play in the risk for disease?
- How do the different causal variants in each cell type contribute to disease? How do these functions interact with other risk factors for MS?
- How do these findings apply to primary progressive MS?
- Does the chromatin landscape of genetic risk factor change for different forms of MS and implicate different enhancers or different cells?
Disclosures and sources of funding
The research was supported by the NIH Common Fund, the National Human Genome Research Institute, the National Institute of Allergy and Infectious Diseases, the National Institute of Neurological Disorders and Stroke, the National Institute of General Medical Sciences, the National Multiple Sclerosis Society, the UCSF Sandler Fellowship, a gift from Jake Aronov, the Penates Foundation, the Nancy Taylor Foundation, and the Howard Hughes Medical Institute.