Altered Immunity, Crippled Neurons
Widespread physiological malfunctions underlie MS
Putting out a fire stops a house from burning, but if the underlying cause is faulty wiring or a habit of smoking in bed, the fix is temporary. A similar situation faces scientists who are tackling MS, the most common cause of neurological disability in young people. The current crop of medications can douse symptoms and slow MS from turning into an inferno, but the events that cause the hallmarks of the disease—disintegrating myelin and deteriorating neurons—remain elusive. Getting to the root of the problem is essential to improving the outlook for MS patients.
“We know that both inflammation and neurodegeneration are critical components” of MS, says Bruce Trapp, a neuroscientist at the Lerner Research Institute of the Cleveland Clinic in Ohio, but it’s not clear which comes first. “One of the biggest questions in MS is whether it is a primary inflammatory disease or a primary neurodegenerative disease,” he says. “We don’t know the answer to that” (Trapp et al., 2008).
Multiple genetic and environmental factors conspire to ignite MS (see "Whodunit?" and "Genetic Associations"; Ramagopalan et al., 2010; Compston and Coles, 2008), but no clear trigger has been established. Although the malady is commonly categorized as an autoimmune disease, no inciting autoantigen has yet emerged. Researchers have concentrated on the immunological features of MS and on the inflammatory roles of the molecules involved in MS, in large part because blood—through which immune cells course—is easier to access than nervous system tissue. Many of the cellular processes that orchestrate MS are already in full swing by the time the first symptoms appear, a situation that obscures the illness’s initial stages. “Our entry point into this disease is still very problematic in terms of what the core, early steps are,” says Trevor Kilpatrick, a neurologist at the University of Melbourne in Australia.
Like an ugly merger of two megacorporations, MS results from clashes between a pair of intricate networks, the immune system and the nervous system, each with its own perplexing org chart. Synthesizing all the parts into a coherent view of the disease is difficult, if not impossible. But researchers are steadily constructing a framework to understand what goes wrong. Building on what they know about normal neuron function, scientists are describing the gross damage that MS does to the brain, how the immune system attacks myelin, and how cellular processes go awry and injure or kill neurons. Insights into these aspects of MS pathology are suggesting new strategies to help those afflicted by the disease.
Rapid transit: normal neural function
Ordinarily, the nervous system is a reliable, efficient network, zipping electrical impulses around the body to control everything we do: eat, breathe, think, pipet, and write grant proposals. Information hops from one neuron to another through synapses, short gaps bridged by chemical or electrical signals. But it’s the movement of electrical current down an axon—the cable that sends information long-distance through the nervous system—that propels information long distances through the nervous system.
Current flows along a neuron’s axon like water cascading down a river after breaching a dam; events called action potentials provide the surge that overwhelms the dam. Channels in the neuron’s cell membrane allow ions such as sodium, potassium, and calcium to cross. These conduits open and close in response to chemicals, changes in membrane voltage, or other signals. When a signal of sufficient strength hits a neuron, sodium channels open and flood the cell with the ion, generating a voltage spike, or action potential, across the membrane. This spike spurs another by prodding channels in the next section of the axon to open. In this way, the voltage spike tumbles down the axon.
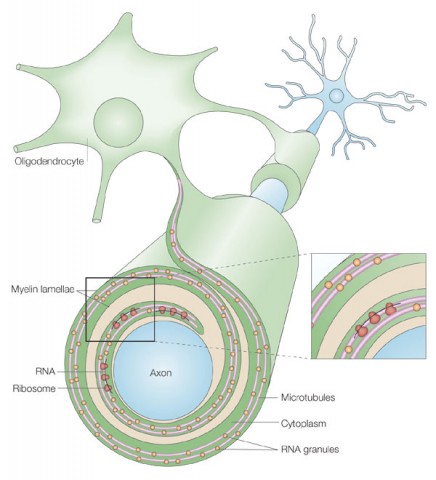
Channels also provide a route through which current can leak. In household electrical wire, insulation protects against such seepage, which would break circuits and create fire hazards. Neurons have insulation, too. Support brain cells called oligodendrocytes wrap around neurons and embrace axons with extensions of their membranes. These membranous casings are called myelin. Channels congregate at the short gaps, or nodes, between sections of myelin. As a result, electrical current jumps from node to node, and the channels at each one provide new oomph to keep the action potentials going.
Surveying the damage
When myelin degrades in multiple sclerosis or other diseases, the transmission short-circuits. Ions leach out as voltage spikes travel down an axon, and action potentials grow sluggish, weakening and slowing the message. Without insulation, signals from neighboring neurons interfere like nearby radio antennas.
MS spares no part of the nervous system. White matter in the brain and the long axons of the spinal cord is replete with myelin. In the traditional view, MS was a white matter disease, because conventional imaging and staining techniques picked up conspicuous plaques in that region. But increasing evidence suggests that gray matter, such as the outer layer of the brain, also suffers, even early in the illness (see "More Than Meets the Eye" and "Gate Crashers"; Lucchinetti et al., 2011). The bulbous cell bodies, each carrying a nucleus, make up the bulk of gray matter, and these parts of the neuron aren’t myelinated—but axons, with their myelin coatings, crisscross this part of the brain.
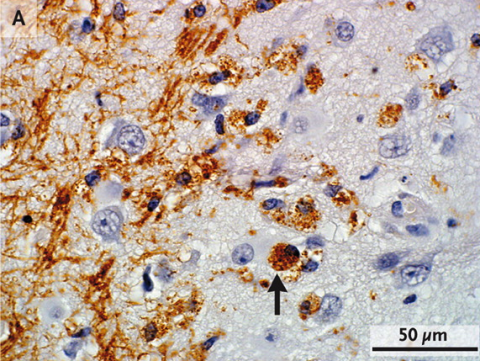
Although MS’s effect on gray matter has been underappreciated, new techniques are revealing that its lesions are present in significant numbers. Furthermore, clinical deterioration tracks more closely with insults to gray matter than white matter (Kutzelnigg et al., 2005). And trouble extends beyond what imaging reveals: Even brain regions without detectable plaques exhibit subtle molecular perturbations that suggest they are either compromised or defending against attack (Zeis et al., 2008; Dutta et al., 2006).
Researchers have learned a great deal by scrutinizing the defects that white matter accrues, but they need to delve deeper into less obvious changes throughout the nervous system, says Nicole Schaeren-Wiemers, a neurobiologist at University Hospital Basel in Switzerland. For instance, iron builds up to an unusual degree in gray matter as MS progresses, yet it’s unclear what role, if any, the metal plays in the pathology of the disease (Williams et al., 2012). Much remains unknown about how pathology varies from person to person, Schaeren-Wiemers adds. “Every brain region is affected and every patient is different,” she says.
At the tissue level, MS wreaks widespread havoc, and no functions of the brain are safe. Blurred vision is often the first sign of the disease. People with MS can develop weak muscles, struggle to move properly, and experience numbness or pain. Tremors, speech problems, and slowed response on memory tasks also occur.
The disease typically strikes first in waves and then in a steady stream. The majority of people with MS—85%—start with so-called relapsing-remitting MS (RRMS), in which cycles of attacks punctuate periods of remission, during which symptoms subside or even disappear. Over time—often after a decade or more—many patients enter into a so-called secondary progressive form, suffering a relentless decline in neurological function with no remissions. A portion of patients—10% to 15%—begin with a progressive form of the disease, skipping the relapsing-remitting stage.
Troublemaking immune cells
In the predominant view of MS, an immune attack on myelin is central to the disease. Even healthy people generate T cells that recognize the body’s own proteins, including myelin. But control mechanisms help prevent these cells from causing harm: Various processes weed them out as they develop or ensure that they don’t spur an immune response. In addition, the blood-brain barrier—a wall between the circulatory system and the central nervous system—prevents immune cells from entering inappropriately and thus buffers myelin from most T cells (see "Gate Crashers").
In multiple sclerosis, however, these self-recognizing immune cells appear to wreak havoc. It isn’t clear why the body doesn’t shut them down, but some clues come from genetics and epidemiology. The most prominent known genetic risk factor is a variant in the major histocompatibility complex, a structure that arranges rendezvous between T cells and the antigens that provoke them into action. Other work has hinted that prior infection by the Epstein-Barr virus (EBV) might instigate trouble (see "Viral Villain"). For instance, some of the protein components of EBV resemble myelin, and they could potentially ignite T cells that recognize myelin and cause those cells to attack the neuronal insulation. Although the disease might start with such a misdirected immune response, an alternative scenario conjures neuron damage from a different—currently unidentified—process that releases bits of myelin into the bloodstream, which then activate T cells. These and potentially other events could foster MS.
Regardless of how the disease begins, numerous lines of investigation have established that altered immunity figures prominently in MS (Lassmann et al., 2007). Perhaps the best evidence comes from existing therapies. “What is clear is that if I give treatments that decrease certain immune processes, it helps,” says Amit Bar-Or, a neurologist at McGill University in Montreal, Canada (and an Accelerated Cure Project scientific adviser). For instance, currently available drugs for MS include natalizumab (Tysabri), an antibody that prevents immune cells from getting into the brain, and forms of β interferon (see interferon β-1a and interferon β-1b), a naturally occurring molecule that reduces inflammation generally, through a mechanism that is not completely clear.
Because these drugs influence multiple parts of the immune system, their effectiveness does not reveal exactly how that system falters in MS. Much of the knowledge about how it goes haywire comes from studies of an animal model called experimental autoimmune encephalomyelitis (EAE). In EAE, animals (commonly rodents, but other species, too) injected with various combinations of myelin proteins develop an autoimmune disease that resembles MS in some ways (see "Animal Arsenal"). Many studies that used this system point to a particular kind of T cell—the helper T cell, also called a CD4+ T cell, named for a surface protein it produces. In the pivotal demonstration that EAE is an autoimmune disease, researchers purified myelin-reactive T cells from an animal with EAE and transferred them to a normal animal; there, they triggered MS-like symptoms such as muscle weakness and paralysis.
That result shows that CD4+ T cells can spark EAE, but it doesn’t address whether CD4+ T cells cause MS. EAE isn’t a perfect MS mimic: The name refers not to a single model but to a set of models, which differ in the myelin components and immunizing techniques they deploy—and none of which replicates all aspects of MS. Furthermore, whether any of these myelin components incite MS in humans remains unclear. Although an immune attack on myelin is central to MS, no one has demonstrated that it is an autoimmune disorder. “[EAE] really isn’t MS,” says neurologist Jerry Wolinsky of the University of Texas Health Science Center at Houston. “It would be an easier disease to study if we had a truly good animal model for the disease. The animal models we have are, frankly, very contrived.”
Recent studies provide additional evidence that EAE doesn’t tell the whole human story. In animals, CD4+T cells seem to be the main actors, but in humans, another type of immune cell may be more important. The telltale lesions seen in the brains of MS patients contain fewer CD4+ T cells than CD8+ T cells, which normally destroy cells that carry viruses and prevent the infectious agents from spreading. In the brains of patients with MS, CD8+ T cells not only outnumber CD4+ T cells, but they also seem to proliferate more rapidly after recognizing antigens in the immune system, such as myelin. No one knows for sure what these observations mean, but—unlike the situation in EAE—it seems plausible that CD8+ rather than CD4+ T cells play a dominant role in MS.
And the focus on T cells of any kind might be leading scientists astray. “If you look back at the therapies that work and those that don’t work, there’s a curious pattern,” says neurologist Timothy Vollmer of the University of Colorado, Denver. Molecules that specifically target T cells fail, he says, whereas every approved therapy acts on other immune players: B cells (Kala et al., 2010). Although many treatments affect T cells too, “there isn’t a therapy approved that doesn’t have B-cell activity.” In other words, effective drugs target many parts of the immune system; those that poke only T cells aren’t helpful.
Bolstering the idea that B cells are central in the disease, studies have demonstrated that a cancer drug called rituximab slows the progression of MS. Rituximab—an antibody that gloms onto CD20, a surface protein found on developing B cells—tags B cells, marking them for destruction (Barun and Bar-Or, 2011). The agent appears to help patients with RRMS and even those who are in the early stages of primary or secondary progressive MS. “In fact, attacking B cells selectively gives you as much benefit as the other therapies currently available,” Vollmer says. “This suggests that B cells are a pivot point in MS.”
B cells are renowned for making antibodies, but rituximab seems to hamper a different B cell function. The drug depletes the cells, yet doesn’t reduce antibody levels because it doesn’t touch the subset of B cells that make antibodies. B cells are also extremely effective at presenting antigens to the immune system—and that activity, as opposed to antibody production, could be the primary way they contribute to MS, Vollmer says.
“When we think about the pathology of the human disease, the therapies that we use inform us about the biology,” Vollmer says. “Now we can go back and test those ideas in animals.” In particular, he says, developing models where B cells play a more prominent role is crucial.
In the end, many cell types likely collaborate to cause MS. The immune system is a tangled web of different types of cells with subtly different functions, and immune responses don’t result from a single action of a single cell type working alone. For instance, once T cells get to neurons, they provoke other immune cells, such as macrophages (immune cells that chew up pathogens) and microglia (resident immune cells that resemble macrophages). Activated macrophages and microglia then produce molecules that kindle inflammation and damage neurons—and they can also generate substances that extinguish inflammation. The field needs to synthesize these individual elements into a comprehensive view of MS origins, Kilpatrick says. Focusing on single cell types “can’t be the way to go,” he says. “There’s got to be a holistic perspective about how these things all interact.”
Resolving issues such as the relative importance of B cells and T cells is difficult enough. MS researchers have also encountered notions that come from far out in left field. In 2008, vascular surgeon Paolo Zamboni of the University of Ferrara in Italy presented a hypothesis that the disease results from constricted blood flow in the veins that drain the central nervous system, a condition that is called chronic cerebrospinal venous insufficiency. He and others have started treating MS patients with angioplasty to open veins. But the idea and the therapy remain controversial. The evidence that MS arises from narrowed veins is thin, and several patients have died or suffered serious side effects from the treatment. Assessments of venous flow are made using ultrasound, and doubts also exist about how well this technique can discriminate between normal and abnormal flow. Furthermore, if veins do narrow in MS, this phenomenon could result from, rather than cause, the disease. Still, some scientists deem the hypothesis worthy of further scrutiny, and more rigorous studies are under way.
Broken neuronal transmissions
Regardless of uncertainty about the particular cellular contributors to the disease, inflammation remains a primary player in RRMS and leads to the characteristic nerve injury. As inflammation waxes and wanes, symptoms emerge and recede in concert, leading to cycles of relapse. But even during this phase of the disease, in which flare-ups subside, irreversible damage occurs. People have enough cognitive reserve to find neuronal detours that allow them to think and move without obvious deficits; up to a point, alternate brain circuits can stand in for damaged ones (Sumowski et al., 2010). “Most humans have [the] ability to shift function from areas of the brain that are injured to areas that are more functional, masking consequence of brain injury,” Vollmer says. Yet, neurons are quietly breaking and dying. The progressive forms of the disease might set in when damage is too severe for the brain to compensate.
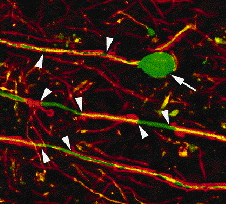
Myelin loss hampers normal transmission of electrical charge down axons, but even worse, it leaves them naked and vulnerable to additional harm. When myelin disappears, the ion channels that control sodium flow across the membrane redistribute themselves. Rather than concentrating at nodes between myelin, they spread out. Furthermore, their numbers increase—particularly the numbers of an ion channel type that doesn’t shut off. As a result, cells bring in an inordinately large amount of sodium. This flood causes another channel to pump in calcium rather than export it as usual. Calcium concentration ramps up inside the cells, inducing destructive activities such as cell-membrane demolition (Waxman, 2006).
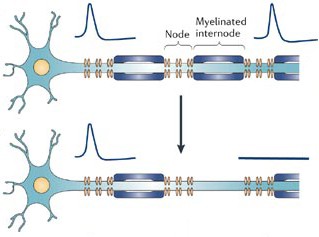
Drugs that shut down sodium channel activity might protect axons directly, by correcting the ion’s levels in neurons. Sodium channel blockers are already approved for other conditions, including epilepsy. Moreover, they can be taken in pill form, thus avoiding the injections required to deliver most current MS treatments. But one sodium channel blocker, lamotrigine, did not produce an improvement for participants in a phase II MS trial (Kapoor et al., 2010). “That was really the first neuroprotective-directed [rather than immune-directed] therapy,” Trapp says. Although the trial failed, new drugs fine-tuned for certain kinds of sodium channels could produce a more promising outcome, he adds. Blocking other types of channels already appears to be an effective strategy, at least for targeting certain symptoms of MS: Dalfampridine, a drug approved by the U.S. Food and Drug Administration 2 years ago that helps MS patients walk more nimbly, jams up potassium channels and aids neuronal conduction.
Other evidence hints that MS disrupts energy balance in brain cells. In animals, EAE smothers the activity of genes essential for the function of mitochondria, organelles that generate the cell’s energy. Analysis of postmortem tissue from human brains suggests that MS similarly pulls the plug on cells (Dutta et al., 2006). At the same time, inflammatory signals from the immune attack on myelin—which starve neurons of oxygen in addition to destroying the insulating sheath—cause mitochondria to malfunction. That perturbation alone could cripple neurons by leaving them without enough steam to cope with problems like a glut of sodium.
Paradoxically, energy blackouts in cells that support neurons can also injure them by making the neurons hyperactive, says Selva Baltan of the Cleveland Clinic (Tekkök et al., 2007). Here, cells called astrocytes play a starring role. Among other functions, they help mop up the neurotransmitter glutamate. Neurons use glutamate to share information; one neuron dispenses the chemical and activates a neighboring neuron. Quantities of the substance must be tightly controlled or neurons can be damaged from overexcitement. If astrocytes are deprived of oxygen, their glutamate pumps run backward, spewing the compound rather than soaking it up. “If we can maintain the energy status of tissue, we will prevent excitotoxicity,” Baltan says. New drugs could help preserve this balance and save neurons. For instance, an agent called riluzole that is currently used for amyotrophic lateral sclerosis, or Lou Gehrig’s disease, blocks glutamate receptors. The compound appears to relieve EAE (Gilgun-Sherki et al., 2003). Researchers are now testing whether it delays neuron damage in patients with early multiple sclerosis.
Prospects for treating early MS are encouraging, but still vexing is how to help those with progressive disease. New views of how the illness evolves could offer some hope. MS shares characteristics with neurodegenerative disorders such as Parkinson and Alzheimer disease, in which substantial loss of neurons occurs many years before people show symptoms (Lassmann, 2011). The similarities raise hope that common themes or approaches could emerge that are helpful for multiple diseases.
Unlike those illnesses, MS affects young people disproportionately. But as MS patients age, they seem to become refractory to anti-inflammatory treatment—at least in the progressive stage. As a result, scientists have supposed that inflammation doesn’t play a role in the progressive form of the disease. “That’s the prevailing impression in the field, but it’s wrong,” Vollmer says. Instead, the blood-brain barrier seems to seal up in progressive MS, trapping active inflammation in the brain and sequestering it from immunotherapies injected into the bloodstream. A 2009 study of autopsy material from MS patients revealed that degree of inflammation and extent of axon damage tracked closely, even in progressive MS (Frischer et al., 2009). In addition, Vollmer says, the body’s immune response changes with age, and the aging process—rather than MS-related events—could limit the effectiveness of anti-inflammatory therapies. In the same study, the oldest progressive subjects exhibited a degree of inflammation similar to that of age-matched controls, reflecting the decline in immune function known to occur in normal aging. That finding suggests that the inflammatory aspect of the disease subsides as people grow older and meshes with a subtle analysis of the rituximab study, Vollmer says. Overall, patients with progressive MS did not respond to the drug, but after separating younger and older progressive patients in the data analysis, researchers found that the younger group did respond—and showed more signs of inflammation beforehand.
The idea that normal aging confounds the evolution of MS adds a layer of complexity when considering how to treat progressive patients. Patients labeled “progressive” aren’t generally given the standard drugs because doctors presume they won’t work. And because progressive patients of all ages have been lumped together in previous studies, those studies support that presumption. “The primary predictor of whether a person responds to a treatment is evidence of inflammation,” Vollmer says. “If you take that 45- or 50-year-old primary progressive patient and don’t treat them, it just doesn’t make sense.”
Damage control
Existing treatments take aim at quelling inflammation, minimizing relapses, and decreasing their severity and frequency. Current drugs have had dramatic success in slowing MS, but stopping or reversing demyelination or axonal damage represents an important new avenue for therapies.
Some research groups are investigating the use of precursor cells—cells slightly more specialized than stem cells—to replenish oligodendrocytes, the brain cells that blanket neurons with myelin. Urging precursor cells to become oligodendrocytes presents the first challenge, but to make a viable treatment, scientists also need to get the cells to the right place and activate their myelination pathways. Any therapy needs to reach the entire central nervous system—the brain and the spinal cord—a requirement that poses a major hurdle. The brain is hard to access, and injection of cells is invasive and difficult to accomplish, especially at multiple sites. But increasing evidence suggests that cells don’t need to be injected into the brain; stem cells infused through the blood can use their natural homing instincts to find their places, raising the promise that cell-based therapies could work in MS (Freedman et al., 2010).
Even if cells get to all the right spots, whether they’ll work properly is unclear. Something about the damaged state in the MS brain seems to prevent oligodendrocytes from making myelin. “It’s not that oligodendrocytes aren’t present in MS plaques,” Wolinsky says. “But they are inhibited from doing what they should do.” Furthermore, the brain should have plenty of regenerating power—without adding more cells. “Five percent of cells in the brain are oligodendrocyte precursors,” Schaeren-Wiemers says. “If we have so many, why don’t they already remyelinate?”
So researchers are trying to identify molecules that could lift these blockades and help oligodendrocytes—whether original or supplemental—make myelin. And some of those molecules are already serving as focal points for new drugs. “The first clinical trial we will have for remyelination is probably an antibody against a molecule called LINGO,” Trapp says. LINGO stunts the maturation of oligodendrocytes, and its quantity escalates in MS plaques. Perhaps jamming LINGO with the antibody will loosen the hold on oligodendrocytes and allow them to produce new myelin around bare axons. Biogen Idec Inc. already has that antibody (called BIIB033) in human safety testing. And thwarting another molecule, death receptor 6, preserves oligodendrocytes and encourages remyelination in animal models (Mi et al., 2011). Biogen Idec is pursuing this molecule as a drug target.
Although that approach seems promising, Vollmer cautions that “MS is a disease of neurons, not of oligodendrocytes.” In the end, malfunctioning neurons rather than misbehaving oligodendrocytes spur the neurological effects typical of MS. Remyelination alone will not rescue neurons that are broken or dead, although it might restore normal communication in neurons that haven’t reached that point. Labs should focus on how to keep neurons healthy and revive those that are sputtering, whether it’s through remyelination or other avenues, he says.
Redoubling attempts to understand how various immune pathways, misbehaving brain cells, and disrupted neuronal signaling pathways coalesce to slay neurons remains crucial to our understanding and future treatments of MS. These efforts, scientists hope, will help clear the smoke that obscures our understanding of the disease. The ongoing quest for this knowledge could help MS patients live longer, healthier lives.
Key open questions
Does a true autoantigen play a central role in MS? If so, what is the identity of this autoantigen?
Are RRMS and progressive MS distinct diseases or two phases of the same disease, and what causes the transition from RRMS to secondary progressive MS?
Can damaged neurons be remyelinated? Is remyelination a practical therapeutic strategy?
What conditions or events cause different parts of the brain to be susceptible in different patients?
How do the environmental, genetic, and epidemiological risk factors link to the specific pathophysiological changes seen in MS?
Image credits
Thumbnail image on landing page and Fig. 1. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience. Mechanisms of axon ensheathment and myelin growth. DL Sherman and PA Brophy, 6: 683-690), copyright 2005.
Thumbnail image on this page. Transmission electron micrograph of a myelinated axon. The myelin layer (concentric) surrounds the axon of a neuron, showing cytoplasmathic organs inside. Generated and deposited into the public domain by the Electron Microscopy Facility at Trinity College. “Myelinated Neuron," 2007 Roadnottaken. Released under a Creative Commons Attribution-ShareAlike 3.0 Unported License CC BY-SA 3.0, as well as under the GNU Free Documentation License.
{kind=link}
Fig. 2. Reprinted by permission from the Massachusetts Medical Society (Inflammatory Cortical Demyelination in Early Multiple Sclerosis. CF Lucchinetti et al., 2011 10.1056/NEJM199801293380502).
Fig. 3. Reprinted by permission from the Massachusetts Medical Society (Axonal Transection in the Lesions of Multiple Sclerosis. BD Trapp et al., 1998 10.1056/NEJM199801293380502).
Fig. 4. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. SG Waxman, 7: 932-941, copyright 2006.