Harnessing Epitope Spreading to Treat MS
Echoes of a vanishing target reveal vulnerability
Like the mythical phoenix that rose from its ashes, a once-hot area of immunology research has returned and may lead to new therapies for MS. “Epitope spreading,” a phenomenon of ephemeral and shifting provocateurs of autoimmunity, was a popular topic of research in the 1990s, but for a variety of reasons funding dried up around 2003. More recently, however, researchers’ interest has been renewed, and some are coming to regard epitope spreading not as a problem in developing MS treatments but as an opportunity.
Burning down the house
In MS, T cells attack components of myelin, including myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP). But the pathology goes beyond an autoimmune attack against a single target.
Epitope spreading is the response to additional epitopes or to other proteins that are part of the target tissue. In MS, T cells react to an initial myelin epitope that an antigen-presenting cell (such as a dendritic cell) displays linked to a particular MHC protein. After symptoms fade, lingering inflammation may send a new wave of not-yet-activated T cells into the CNS, where they become primed to new epitopes (McMahon et al., 2005) in the presence of costimulatory signals. These secondary epitopes may be on the same or different myelin proteins.
 The course of epitope spreading reminds Vincent Tuohy, Ph.D., an immunologist at the Lerner Research Institute at the Cleveland Clinic in Ohio, of a house fire. In the field’s heyday, he led a team that followed epitope spreading in patients with isolated monosymptomatic syndromes that often precede diagnosis of MS (Tuohy et al., 1998).
The course of epitope spreading reminds Vincent Tuohy, Ph.D., an immunologist at the Lerner Research Institute at the Cleveland Clinic in Ohio, of a house fire. In the field’s heyday, he led a team that followed epitope spreading in patients with isolated monosymptomatic syndromes that often precede diagnosis of MS (Tuohy et al., 1998).
“Imagine a house on fire that starts in the kitchen,” Tuohy says. “The kitchen burns down and the fire spreads to the living room and then spreads to the next room. By the time the fire is in the third room, the fire in the kitchen is out because there’s nothing left to burn and only smoldering embers can be detected there. In epitope spreading, inflammation spreads like a fire until every epitope that can elicit a response occurs in a sequential and often predictable cascade, and the whole house burns down.”
But each house fire is different, and so is epitope spreading. “Different patients respond to different antigens, some to MOG, some to MBP, some to PLP. But within those are sequestered epitopes that are often unique to individual patients,” says Don Healey, Ph.D., chief scientific officer of Opexa Therapeutics Inc. in The Woodlands, Texas. And that individuality in response may be the basis of a new, personalized way to treat MS.
Discovery in the early 1990s
The research group of Eli Sercarz, Ph.D., at the University of California, Los Angeles Geffen School of Medicine discovered epitope spreading (Lehmann et al., 1992, 1993).
Thomas Forsthuber, M.D., Ph.D., a professor in the University of Texas at San Antonio department of biology, fondly recalls his time as a postdoctoral researcher in the Sercarz lab: “It was like the Woodstock of science, with the freedom to develop your own projects.” Sercarz’s 2009 obituary describes a connoisseur of exotic cheeses who was an energetic researcher known to suddenly dance about the lab.
Another postdoc, Paul Lehmann, M.D., Ph.D., led the project. He’s the founder, president, and CEO of Cellular Technology Ltd. Lehmann and Forsthuber worked with the experimental autoimmune encephalomyelitis (EAE) mouse model of MS. They injected mice with bits of MBP and 10 days later saw a T cell response from draining lymph nodes. But by day 40, it had vanished. “That’s where we made the leap. Paul Lehmann said, ‘Maybe this can be optimized,’ ” Forsthuber recalls. The two postdocs then discovered a delayed response, in the spleen, where memory T cells became a tool to observe events past day 10.
Not many people were working with the spleen back then, and detecting a memory T cell response was challenging. But Lehmann and Forsthuber found that if they tweaked the culture conditions, replacing fetal calf serum with serum-free medium containing amino acids, memory T cell responses emerged from the background noise.
The two young researchers quickly became known for their finesse with spleens, demonstrating their techniques at scientific meetings in the early 1990s. The ability to watch an autoimmune response unfold past the initial stimulus revealed epitope spreading.
“This was the tool that allowed us to look at T cell memory responses later on. What we did was to immunize with one peptide and wait. We got response to different peptides completely unrelated to the initial immunizing peptide,” Forsthuber recalls. Others showed that if you target those subsequent peptides, you can modulate how diseases behave, he adds. One of those others was Stephen Miller, Ph.D., a professor in the department of microbiology and immunology at the Northwestern University Feinberg School of Medicine in Chicago, Illinois.
The choreography of epitope spreading
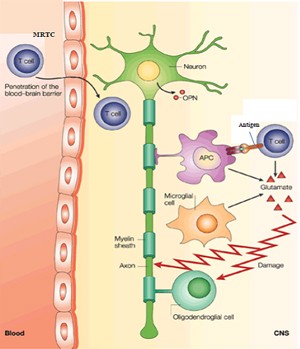
Miller’s group demonstrated the role of epitope spreading in MS pathology (McRae et al., 1995), taking advantage of being able to pinpoint when the initiating event occurs in the mouse model. If they induced immune tolerance against the initiating epitope before the age at which acute symptoms manifest, they could prevent the symptoms. But if they tried to induce tolerance after mice had already exhibited symptoms, it wouldn’t work. The secret turned out to be targeting more than one epitope.
”If we induced tolerance to a mixture of antigens—the spreading epitopes—by administering them during remission, relapses could be ameliorated or prevented. This suggests that the progression of disease is due to immune responses specific for regions of myelin proteins not used to initiate the disease,” Miller explains.
Epitope spreading is dynamic, like the spreading fire. Forsthuber’s unpublished observations of the phenomenon in people support the findings in mice: “At time point zero, a certain set of peptides induces responses. Three months later, some of these peptides don’t induce a response anymore and new peptides do. Later, we see even newer ones, and the older ones are gone.”
Too complex to tackle?
A disappearing target that gives rise to additional targets that vary among individuals is not exactly a pharmaceutical company’s dream. But it fits right in with personalized medicine.
“I remember going to drug companies showing data on how to induce tolerance to several MBP and PLP peptides after disease induction, and they didn’t want to hear that it’s not just one peptide, it’s a whole array of peptides that can be targeted,” Tuohy recalls.
Where pharma saw epitope spreading as an unexpected complication, Tuohy and others saw opportunity. “Yes, the disease was more complex than we thought. But instead of thinking it didn’t provide a very distinct regimented single epitope to tolerize against, which is what many anticipated and wanted, it provided several peptides that one could tolerize against.”
Tuohy returns to the fire metaphor. “All you have to do is jump ahead of the fire. Put ice in the other rooms. Hose them down. You don’t have to hit every peptide. You have to tolerize against something to slow the disease down. Aim your flame retardant at a room that hasn’t yet burned down.”
Funding woes
The reasons for the decrease in U.S. government funding for epitope spreading that began by 2003 remain murky. Part of the problem is that some experimental results couldn’t be repeated. Tuohy explains why. “We found that if the initial symptoms weren’t severe, detecting epitope spreading was less likely. I think this may be why some investigators observed it and some didn’t.”
Or declining interest could have simply represented the fading of a fad. Suppressor T cells, interferon beta, monoclonal antibodies—they’ve all been the darlings of immunology research. “Twenty years ago, a grant proposal to discover new T cell epitopes was fundable. That’s no longer so. Reviewers for funding agencies want new and sexy things,” Forsthuber says.
Treatment based on epitope spreading might be more precisely targeted than the existing approved drugs for MS, which are broad-based and cause unwanted immunosuppression. “Let’s prevent T cells from going to the brain! Let’s target every T cell! None of the FDA-approved treatments are specific for pathogenic T cells,” Forsthuber says. But selectively vanquishing specific sets of T cells, especially memory cells, is difficult. “It’s much easier to wipe out half of the immune system or prevent cells from getting into the brain or proliferating,” he adds.
Two promising therapeutic approaches based on epitope spreading in clinical trials are funded from outside the NIH. Miller’s group, along with that of Roland Martin, M.D., of the Center for Molecular Neurobiology in Hamburg, Germany, is funded mostly by the German government, the Cumming Foundation, and the Myelin Repair Foundation. The other, more personalized, experimental treatment is from Opexa Therapeutics.
Tolerance to shared epitopes
Miller and Martin’s phase 1 clinical trial evaluated the safety of autologous leukocytes bearing bits of the three major myelin proteins (Lutterotti et al., 2013). The approach builds on an old observation on mice: Crosslinking peptide antigens to leukocytes induces apoptosis. “Injecting antigen-coupled leukocytes intravenously induces immune tolerance,” Miller explains. “The way it works is that dead and dying cells are filtered out in the liver and spleen, which contain cells that present the antigens to T cells in a way that rather than activating them makes them tolerant.”
Martin’s group had identified seven peptides that are likely those that emerge in epitope spreading. The researchers attached these to leukocytes from seven people with relapsing-remitting MS and two with secondary progressive MS. The cells were separated with leukophoresis and up to 3 billion per patient covalently linked to the seven antigens and then infused. As a control, the investigators tracked response to tetanus epitopes. The patients developed tolerance to four of the seven myelin epitopes, and not, as expected, to the tetanus antigens. But even as Miller is planning the larger phase 2 trial, he’s thinking ahead.
A recent paper in Nature Biotechnology outlines Miller’s new approach: biodegradable nanoparticles made of poly(lactide-co-glycolide) bearing encephalitogenic peptides that have prevented and treated EAE in mice (Getts et al., 2012). “The expense and complexity of hooking up a patient to leukophoresis could only be done at large medical centers,” Miller says. “So we looked for surrogate ways to attach antigens that would negate the necessity to inject apoptotic cells.” Furthermore, any antigens could ride on the nanoparticles, to treat different autoimmune conditions. “And it’s all directly related to epitope spreading,” he adds. The NIH, Myelin Repair Foundation, Juvenile Diabetes Research Foundation, and the Australian government funded the study.
A personalized approach
Opexa Therapeutics is using epitope spreading by pitting T cells against T cells. One product, Tcelna, consists of myelin-reactive T cells from the peripheral circulation that are expanded, irradiated, and infused back into the patient, where they raise an immune response against the T cells that drive the disease. The idea comes from the individuality of patients’ responses to secondary epitopes. “In January a patient may respond to MOG, but by the end of the year respond to PLP or to different epitopes within them. By reviewing the immune response to myelin annually, utilizing a peptide library encompassing the three major myelin antigens, we can personalize our T-cell immunotherapy to those responses that are most relevant and likely to be the primary drivers of MS in each patient,” Healey says.
The company has conducted five clinical trials, giving Tcelna to 144 patients in phase 1 and phase 2b studies for relapsing-remitting MS. An ongoing placebo-controlled phase 2b clinical trial (Abili-T) for secondary progressive MS will enroll 180 patients. So far, in the most aggressive cases, the researchers find a reduction in relapse rate with evidence for stabilization in disease progression—without the global immunosuppression and adverse effects of existing treatments.
These recent and ongoing clinical trials, and continuing work in the mouse model, may put epitope spreading back on the map. Sums up Tuohy, “Most scientists agree, epitope spreading is a very real, pathogenic autoimmune process. It seems to be a plastic, vibrant part of the immune response that could help explain the chronic nature of MS and the rejuvenation of autoreactivity as time goes on.”
Key open questions
- How long do pathogenic T cells live?
- How similar is the sequence of unveiling of cryptic epitopes among individuals?
- Can we predict which epitopes will become dominant as the pathology progresses?
- Does epitope spreading happen in all people with MS?
- What are the genetic polymorphisms, in addition to MHC type, and what are the environmental triggers that contribute to susceptibility to MS?
Comments
Lots of epitope spreading research was done in the 90's in animal models. My opinion is that there was good evidence that epitope spreading has an important role in disease in these animal models and stopping these new immune targets could stop disease. In humans that is not so easy to "prove". But If you look at Steve Miller's work, he is now using his "tolerance" methods in human trials. In animal models we could inactivate autoreactive T cells specifically and leave the bulk of the immune response intact to fight infection. Steve is now doing this in humans, but using the concept of epitope spreading he targets multiple antigens.