Animal Arsenal: An overview of animal models that mimic key aspects of MS
“All models are wrong, but some are useful,” British statistician George E. P. Box wrote 35 years ago. Although he was referring to calculations regarding the interaction between two variables, the observation applies equally well to experimental animal systems for multiple sclerosis.
Researchers have gleaned many insights about the clinical and biological nature of MS from studies of patients and autopsy tissue. But model organisms or systems in which some features of the disease have been induced—primarily rats, mice, rabbits, guinea pigs, and nonhuman primates such as marmosets and rhesus monkeys, but also sheep, dogs, and fish, as well as cultured cells—serve as indispensable tools for taking a closer look at cellular mechanisms that underlie its pathology. In addition to serving as a testing arena for potential therapies, animal models are a relatively convenient source of tissue from the central nervous system (CNS), which is the main target of MS.
In a perfect world, an animal model for putative autoimmune diseases such as MS should have the following features, according to a 2012 review (Wekerle et al., 2012): First, it should reflect most or all aspects of the disease; second, symptoms should occur spontaneously rather than be artificially induced; third, the animals should experience illness in the same relative time frame as humans do; fourth, the cellular pathogenesis of the model should bear a strong resemblance to what’s known about human disease; finally, it should not make assumptions about which antigens cause the trouble.
MS models generally fail on most or all of these counts. All established experimental versions of the malady are induced or engineered, and because no one knows what sparks MS in humans, researchers can’t set off an “authentic” trigger. But even if the right trigger were used, there’s no guarantee that the pathological features in animals would reflect those in humans. In addition, the time frame differs: In humans, physiological processes underlying the disease probably smolder undetected for years before problems arise, but symptoms in the animal models surface within weeks or even days after induction. Such acute onset is likely to have a different mechanism than that of MS. To complicate matters further, the fact that the disease manifests itself so differently among individual patients suggests that no single experimental system could capture all—or even most—of its features.
Some scientists warn that the imperfections of animal experiments, especially when used for testing novel compounds, can and do breed results that misinform the field. “Models are good when you know what questions to ask,” says Vijay Kuchroo, a neurologist who studies autoimmune disease at Harvard Medical School in Boston. “They can help you, or they can lead you the wrong way.”
Such limitations haven’t stopped researchers from trying. Animal models for MS have proliferated, and the wealth of options provides an opportunity to dissect the disease's different facets and investigate the mechanisms that underlie its multiple components. Although no single model fully recapitulates MS, each one reflects some characteristic of it. The many possibilities are “a really positive aspect” of the current situation in the field, says Brian Popko, a molecular geneticist at the University of Chicago. “We’re able to use these different models for different reasons to selectively ask questions.”
Experimental autoimmune encephalomyelitis
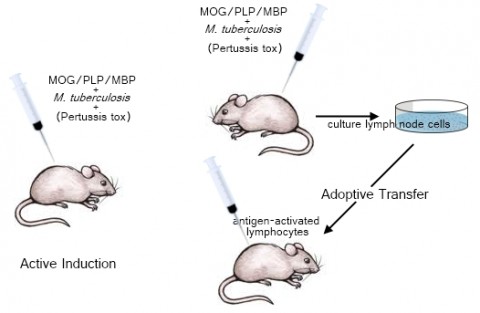
Experimental autoimmune encephalomyelitis (EAE) is one of the oldest models of neurological disease, according to Richard Ransohoff, a neurologist at the Cleveland Clinic in Ohio (and an MSDF scientific adviser). It was discovered in 1933 by researchers who were studying the neurological effects of rabies, and it came on the scene as a system for investigating human demyelinating disease in 1947 (Wolf et al., 1947). Most experimenters induce EAE in rats or mice; its hallmark is the ability to stimulate a T-cell-mediated immune response, but the precise pathology depends strongly on the animal strain. Indeed, it’s more a collection of models than a single one. The disease can be instigated in one of two ways. In active induction, researchers inject animals with an antigen—often a chunk of a myelin protein—along with an emulsified solution that contains heat-killed Mycobacterium tuberculosis, which boosts the proinflammatory response; some mouse strains also require a shot of pertussis toxin, which promotes EAE possibly by facilitating T cells’ access to the CNS. Adoptive transfer, on the other hand, involves priming a donor animal with one of the antigens, harvesting and culturing its activated T cells, and then injecting them along with an immunostimulant into another animal to induce disease (Miller and Karpus, 2007). “They really have fundamental differences,” Miller says. Adoptive transfer more closely reflects human MS, he explains, because active induction “is complicated by the fact that antigen is being released literally over months and months from the site where it is deposited.”
Most researchers agree that EAE has propelled major advances in knowledge about basic immunology as well as the immunological elements that underlie MS (Croxford et al., 2011; Pachner, 2011). But appraisals of its utility in drug development are more varied. The most convincing example of its success is natalizumab (Tysabri), a potent immunomodulator that emerged from EAE experiments in Lawrence Steinman’s lab at Stanford University in Palo Alto, California, 2 decades ago (Yednock et al., 1992). Researchers credit EAE as the cradle of discovery for a handful of other treatments, namely glatiramer acetate, fingolimod, and mitoxantrone, and they acknowledge that the model has helped elucidate the mechanisms of drugs that were identified by other means. Steinman co-authored an EAE-based study in 2010, for example, that explains why interferon β (interferon β-1a and interferon β-1b), the most widely used treatment in MS, doesn’t work in one-third of the patients who take it, and lays the groundwork for a test that might identify people who will not respond (Axtel et al., 2010).
However, the last few years in particular have brought out a clamor of complaints about this model’s use in drug discovery. “[EAE] is worthless for testing drugs, and it’s critical and invaluable for basic research,” Ransohoff says. “We don’t know enough about the fundamental pathogenesis of every EAE model” to guess whether a substance that effectively fights EAE would behave similarly in MS. A compound that blocks tumor necrosis factor, for instance, seems to ameliorate EAE in mice but exacerbates disease in humans (The Lenercept Multiple Sclerosis Study Group, 1999). And gumming up the works with false-positive tests is only part of the problem, he stresses: “You just never know what is sitting up there on the shelf that nobody will ever look at because it failed the EAE experiment.”
Sloppy use of the model worsens the bad biological matchup between EAE and MS, critics say. Researchers commonly administer their experimental treatment before inducing EAE, says David Baker, a neuroimmunologist at Queen Mary, University of London. That means they are asking the drug to prevent inflammation from starting—a much easier task, and much less relevant to human disease, than stopping it after it has begun. An analysis of more than 1000 studies about preclinical models of MS by an Edinburgh University team found this trend in about two-thirds of all EAE studies (Vesterinen et al., 2010). “If you go back and look at how [researchers testing therapies] did the experiments, it’s often in prevention mode,” Kuchroo says. “If you do the treatment mode, there are not very many that work.”
Timing of induction isn’t the only problem. “There are lots and lots of subtleties that are very commonly overlooked,” Ransohoff says. The model is all too often “a go/no-go screen without a lot of investigation of what happened,” he says.
The field is starting to give ear to these problems, however—and more importantly, to voice possible solutions. In a paper published in November 2011, Baker and his colleague Sandra Amor at the VU University Medical Center in Amsterdam present guidelines for writing papers about research that uses EAE and for reviewing them (Baker and Amor, 2011). The idea, according to the duo, is “to constructively improve the quality of published work and reduce the futile use of animals in research.”
Viral and toxicological models
Viral infections, compounded by genetic susceptibility and other risk factors, have long been on the list of possible triggers for MS (see "Whodunit?"and "Viral Villain"). Although researchers have not pinpointed a specific viral culprit, some mouse viruses cause classic MS-like symptoms such as T-cell-mediated inflammation and CNS demyelination. Injecting these viruses into animals has shed light on the possible role of viral infection in MS-like inflammation, how a CNS-persistent virus might trigger autoimmunity to myelin proteins, and basic properties of demyelination, says Stephen Miller, an immunologist at Northwestern University Medical School in Chicago, Illinois.
The different viruses—most commonly, Theiler’s murine encephalomyelitis virus and mouse hepatitis virus (MHV)—initially produce distinct disease states. Theiler’s virus, in its acute phase, causes a motor neuron disease similar to amyotrophic lateral sclerosis, or perhaps polio, says Ehud Lavi, a neuroimmunologist at Weill Cornell Medical College in New York City, whereas MHV at first resembles encephalitis and hepatitis. After about 3 weeks with either infection, some animals enter a chronic demyelinating stage. “Neither one of these models actually mimics what happens in humans, but each one of them can give some clues about the interaction between viruses and the central nervous system, and viruses and autoimmunity,” Lavi says. Researchers are also uncovering additional viruses that might prove useful for studying MS, including one that naturally infects monkeys (Axthelm et al., 2011).
Models that use toxic agents to elicit demyelination in rodents arguably differ more strongly from MS than viral and EAE models do; still, investigators can use them to study fundamental aspects of the process, some of which might apply not just to MS but also to other demyelinating disorders. One commonly used toxin is cuprizone, a copper chelator that is mixed into rodents’ chow. Mice begin to experience demyelination, primarily in the corpus callosum, a band of white matter that connects the two hemispheres of the brain, about 3 weeks after they begin ingesting it, and the damage reverses within a few weeks after they stop. Other toxins, such as lysolecithin and ethidium bromide, are injected into the spinal cord or the brain. Those substances begin to take effect within days. Toxicological models are valuable specifically because they don't contain an autoimmune component, says Wendy Macklin, a neurobiologist at the University of Colorado School of Medicine in Denver. That characteristic means that researchers can observe how nerve cells remyelinate as they naturally do to some extent after an MS relapse—without the additional variable of inflammation—and can investigate interventions that might shore up that process.
Models 2.0
Immunologists were quick to take advantage of gene-targeting technology after it was invented in the late 1980s, and by the next decade, their ability to tinker with mouse genetics began to yield a new crop of animal models for MS. One approach involves creating so-called humanized mice by weaving in elements of the human immune system—for example, certain versions of human leukocyte antigen genes, which represent the major genetic risk factor for MS (see "Genetic Associations"). Researchers also routinely engineer mice that produce T cell receptors for specific antigens—generally the same myelin proteins that are used to incite EAE. Animals that lack genes that encode cytokines or other immune molecules have also provided valuable data. In one especially promising model, researchers crossed two lines of genetically engineered mice to produce a strain that manufactures both T cell and B cell receptors that recognize a single myelin protein (Krishnamoorthy et al., 2006; Bettelli et al., 2006). About half of the animals spontaneously develop EAE, making it the first experimental system for MS in which animals get sick without further human intervention. But even when EAE must be induced in these genetic mutants, the resulting animals might better reflect human illness than does classic EAE in normal mice; furthermore, by testing mice with particular genetic alterations, investigators can assess whether and how certain genes contribute to symptoms.
Animals other than rodents are also gaining ground. Zebrafish, for instance, are beginning to aid studies of new demyelinating agents (Buckley et al., 2010). These organisms are inexpensive, fast-breeding, and transparent in the early stages of the organism's development, features that allow investigators to observe their neurons in vivo.
Researchers are also developing and improving cell-culture systems to study specific aspects of the disease—for instance, how immune cells move through the human blood-brain barrier (Man et al., 2012, and "Gate Crashers").
What’s really needed, experts agree, is a model for the progressive form of the disease, which is marked by steady degeneration rather than the transient attacks that characterize the relapsing-remitting form; a handful of drugs have emerged to treat the latter type of illness, but devising treatment for the former has proved an intractable challenge. “There, the field becomes really constricted and narrow,” Ransohoff says. “We have very few people working on it, and no established animal models at all.” Baker’s group has used a protocol that snuffs out relapses in mice with EAE, thus allowing the rodents’ conditions to decline continuously as it does in progressive MS (Pryce et al., 2005), but he says the 6 months it takes for the animals to reach that state makes drug testing onerous. One of the hallmarks of progressive MS is neurodegeneration, which EAE is not well equipped to replicate, Baker says, but because EAE is relatively straightforward to induce and because it is so widespread, drugmakers are overly fixated on its use. “I think that’s one of the problems,” he says. ”Companies need to see data in an EAE type of model.”
Meanwhile, the recent flood of data about the genetics of the disease in humans provides a new wave of opportunity for understanding the basic biology of the disease, Kuchroo says. By creating mice that carry combinations of these human gene variants—several that act in the same molecular pathway, for example—researchers can begin to tease apart how the genes contribute to MS pathology, he notes: “That will be the next frontier.”
Image Credits
Fig. 1. Alla Katsnelson.
Fig. 2. "Danio rerio (zebrafish) embryonic development," Steve Baskauf, Copyright 2002.
Suggested by Teruna J. Siahaan
This article describes the importance of alpha4-beta1 integrin on leukocyte infiltration through the blood-brain barrier (BBB). This finding was used as the basic principle to develop natalizumab (Tysabri®) for treating MS patients. Natalizumab blocks leukocyte adhesion to vascular endothelium of the BBB and prevents infiltration of leukocytes into the brain.